
еЊвЊ ЖдЛЗыФНјааСЫЗжРр, ВЂЖдОпДњБэадЕФОЕфаЭЁЂDKP (Diketopiperazine, ппрКЖўЭЊ)аЭЁЂУбЧХаЭЁЂЯЉЧХаЭЁЂЕЅСђМАЖрСђУбаЭЁЂИеадЧХаЭЁЂжй(Ъх)АЗаЭЁЂMannich МюаЭЁЂСЊБНаЭМА Freidinger аЭЛЗыФЕФКЯГЩЗНЗЈж№вЛНјааСЫУшЪі.
НќМИЪЎФъРДДгздШЛНчЁЂЛњЬхФкЛђЛЏбЇКЯГЩЕУЕНЕФДѓСПыФЛЏКЯЮя, вдИпЩњЮяЛюадЁЂЕЭЖОИБзїгУЕФЬиЕув§Ц№СЫвЉЮяЛЏбЇМвЕФИпЖШЙизЂ. аэЖрЧщПіЯТ, жБСДыФЕФЗжзгШсЧњад(flexibility)дьГЩЕФЙЙЯѓвзБфЪЙЦфгыЪмЬхНсКЯЕФЧПЖШМАбЁдёадЪмЕНгАЯь. ДЫЭт, ЛњЬхФкЕФАБыФУИМАєШыФУИвВКмЗНБуЕиДгжБСДыФСНИіЖЫЛљж№ВНЧаИюыФСД, ЪЙжЎНЕНт. вђДЫыФСДЕФЛЗЛЏИФдь, ЪЙЦфЙЙЯѓЯоЖЈ(constrained conformation)ЪЧИФЩЦыФЗжзгЩњЮяЮШЖЈадЁЂЬсИпЩњЮяЛюадЕФРэЯыЭООЖжЎвЛ. ВЛЩйбаОПвбОжЄУї, ДгжБСДЕФЯШЕМНсЙЙыФИФЮЊЛЗыФКѓЪЙдгаЕФЩњЮяЛюадЬсИпЪЎМИБЖжСМИЭђБЖ. аэЖрОпгаПЙОњЁЂПЙВЁЖОЁЂПЙжзСіЁЂУтвпЕїНкЕШЛюадЕФЬьШЛВњЮяыФЭљЭљКЌгаВЛЭЌРраЭЕФжїСДЛЗЛЏНсЙЙ. БОЮФФтДгЛЗыФЕФЯЕЭГЗжРрШыЪж, ЖдвЛаЉСДЛЗ(loop)жаВЛЭЌЧХСЌНсЙЙЕФЛЗыФЬиеїМАКЯГЩЗНЗЈНјаазлЪі.
1 ЛЗыФЗжРр
ШчКЮЖддкздШЛНчЦ№зХживЊзїгУЕФЛЗыФНјааЗжРр, жСНёЩаЮовЛИіШЈЭўЕФЁЂбЯИёЕФБъзМ. БОЮФДгвдЯТМИИіВЛЭЌЕФНЧЖШНјааЙщРр:
1.1 ДгѕЃАЗМќНЧЖШЗжРр
ДгѕЃАЗМќНЧЖШПЩНЋЛЗыФЗжЮЊОљЛЗыФ(Homodetic cyclopeptides)КЭдгЛЗыФ(Heterodetic cyclopeptides)СНРр. ОљЛЗыФжИжїСДЛЗОљгЩѕЃАЗМќСЌНг, вђДЫЧХСЌНсЙЙЕЅвЛ; дгЛЗыФжИжїСДЛЗжаГ§СЫѕЃАЗМќЭт, ЛЙгаЦфЫќЧХСЌНсЙЙ, вђДЫгжАќРЈЖржжРраЭ.
1.2 АДРДдДЗжРр
ДгРДдДНЧЖШПЩАДЭМ 1 ЖдЛЗыФНјааЗжРр:
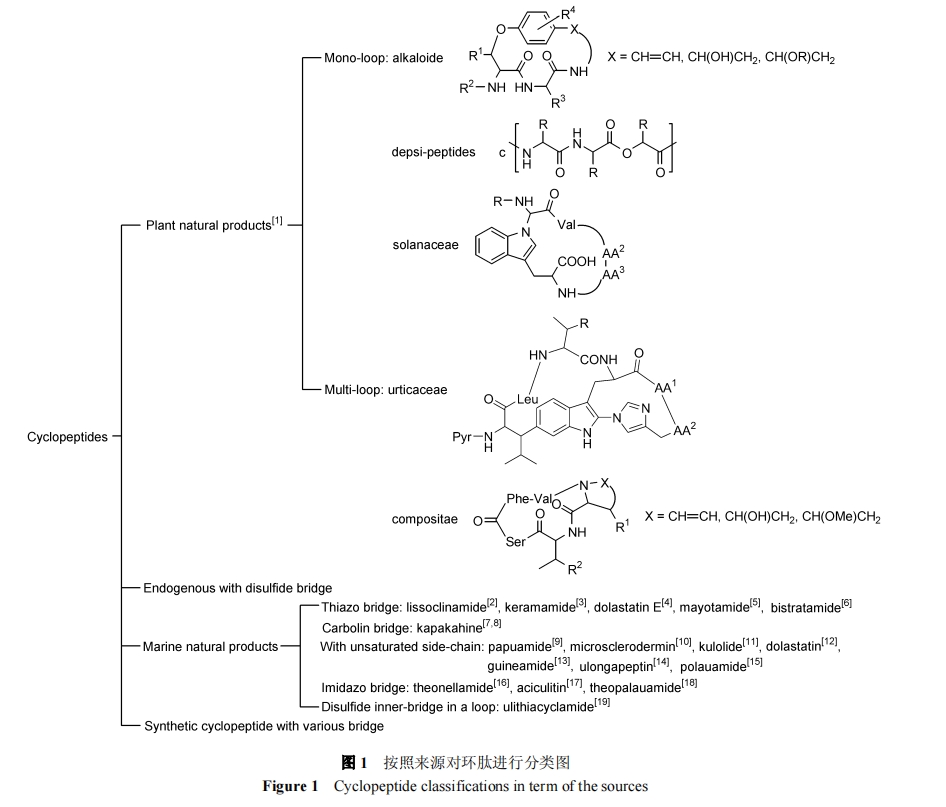
1.3 АДЧХЭЗЮЛжУЗжРр
АДЧХЭЗЮЛжУПЩНЋЛЗыФЗжЮЊ 7 Рр, МћЭМ 2.
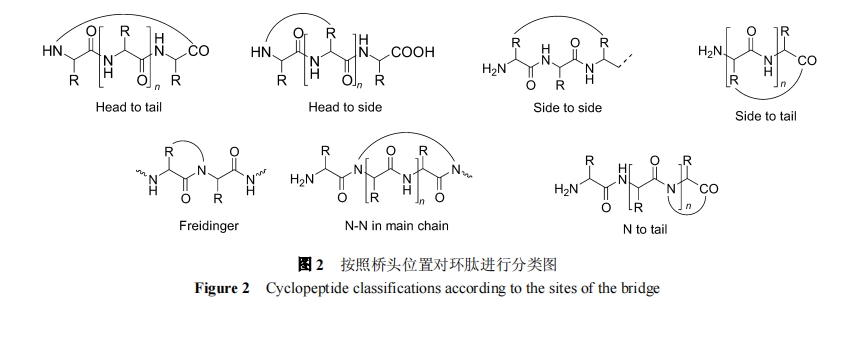
1.4 АДЧХСЌНсЙЙЗжРр
АДЧХСЌНсЙЙПЩНЋЛЗыФЗжЮЊОЕфЛЗыФКЭЗЧОЕфЛЗыФОЕфЛЗыФгжАќРЈФкѕЃАЗЧХЁЂФкѕЅЧХКЭЖўСђЧХ; ЗЧОЕфЛЗыФАќРЈУбЧХЁЂЯЉЧХЁЂЕЅСђЛђЖрСђЧХЁЂИеаджЇМмЧХЁЂАЗЧХЁЂСЊБНЧХЁЂMannich МюЧХЁЂPNA (Peptide nucleic acid, ыФКЫЫс)ЧХЕШ.
1.5 АДжїСДЛЗЪ§ЗжРр
АДжїСДЛЗЪ§НЋЛЗыФЗжЮЊЕЅ loop аЭ, ЫЋ loop аЭКЭЖрloop аЭ.
2 ОЕфЛЗыФКЯГЩ
вбгаЕФИїжжЛЗыФЛЏКЯЮяжа, вдФкѕЃАЗ(гжГЦОљЛЗыФ)ЁЂЖўСђМќЛђФкѕЅМќ(depsi-)ЮЊЧХСЌНсЙЙЕФЛЗыФеМОјДѓЖрЪ§, ЖјЧвЫќУЧЕФКЯГЩЗНЗЈБШНЯГЃМћЁЂОЕф. вђДЫетШ§жжЛЗыФгжБЛГЦЮЊОЕфЛЗыФ.
2.1 ФкѕЃАЗЮЊЧХЕФЛЗыФКЯГЩ
ДЫРрЛЗыФЕФКЯГЩР§КмЖр, змЬхЩЯПЩвдДцдкСНИіЗНЪН: (1)ЪЙгУЫѕКЯМСЕФєШЛљЛюЛЏаЭ; (2)ЮоЫѕКЯМСЕФЗжзгФкАБНтаЭ. ЧАепЖдОљЯрШмвКЗЈМАЙЬЯрЗЈЖМЪЪгУ, КѓепЖргУгкЙЬЯрКЯГЩ. вбгаЮФЯзМЧдиЕФФкѕЃАЗЛЗыФКЯГЩЗЧГЃЖр, ЯТУцНіНщЩмвЛаЉгаДњБэадЕФЗНЗЈ.
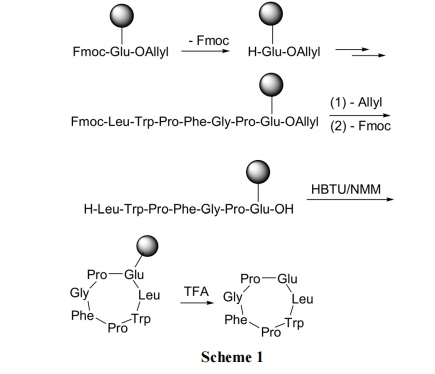
2.1.1 ВрСДСЌНгЙЬЯрЗЈ
РћгУЙШАБЫс(ЛђЬьЖЌАБЫс)ЕФВрСДCOOHгыЙЬЯрдиЬхаЮГЩѕЅМќСЌНг. ЫцКѓЯШЭбГ§ Fmoc, ЪЙЙШАБЫсЕФ ІС-АБЛљгЮРы, вдБуЯђ N ЖЫзщзАыФСД. ШЋВПЫѕКЯЭъГЩКѓ, гУюйЪдМСЭбГ§ C ЖЫ Allyl, ЪЙЙШАБЫсЕФєШЛљгЮРы; дйЭбГ§ N ЖЫЕФFmoc, ЪЙыФСДNЖЫАБЛљгЮРы. зюКѓдкЫѕКЯМСHBTUзїгУЯТ, N ЖЫМА C ЖЫЫѕКЯаЮГЩФкѕЃАЗМќаЮГЩЛЗыФ(Scheme 1)[20].
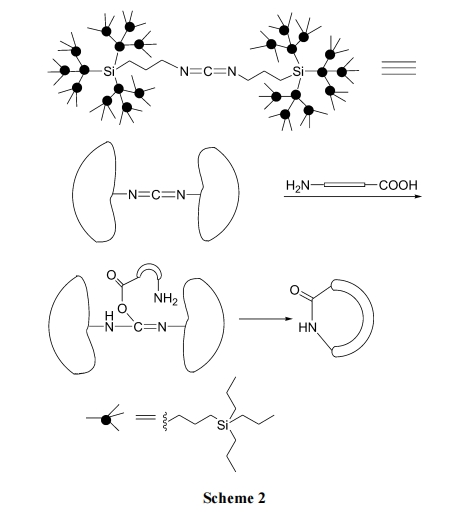
2.1.2 ШмвКМйЯЁЪЭЗЈ
дкДЋЭГШмвКЗЈНјааЗжзгФкЛЗКЯЗДгІЪБ, ЮЊСЫБмУтЗжзгМфЕФЫѕКЯБиашЪЙЗДгІЮядкШмвКжаОЁСПЯЁЪЭ. ШЛЖјетжжИпЖШЯЁЪЭЕФЬѕМўЛсдьГЩЗДгІЪБМфбгГЄЁЂИБЗДгІНЯЖр, вВИјКѓДІРэДјРДВЛЗНБу. еыЖдетжжЧщПі, Amore ЕШ[21]ЪЙгУвЛжжПеМфЮЛзшДѓЕФЪїЭЛзДЙшЭщШЁДњЕФЬМЖўбЧАЗзїЮЊЫѕКЯ МС , Дњ Ьц ГЃ Йц Ъд МС DCC (N,N-dicyclohexylcarbodiimide, ЖўЛЗМКЛљЬМЖўбЧАЗ)НјааШмвКЗНЪНЕФФкѕЃАЗЛЗыФКЯГЩ, ЕУЕНДПЖШКмИпЕФФПБъВњЮя(Scheme 2).
2.1.3 ЙЬЯрЗжзгФкАБНтЗЈ
гЩгкєШЫсыПѕЅ(RCOONЃНR')МќЖдЧзКЫЪдМСИпЖШУєИа, ПЩвдЪЙгУвдыПЮЊ linker ЕФЙЬЯрдиЬх, жЦБИЭЗ-ЮВМќКЯЕФФкѕЃАЗЛЗыФ. зюКѓвЛВНЛЗКЯВЛгУЫѕКЯМСПЩвдЭЌЪБЭъГЩЗжзгФкАБНтМАЭбГ§ЙЬЯрдиЬх(Scheme 3)[22].

2.1.4 ЙЬЯрБЃЯеСЌНгаЭЗжзгФкАБНтЗЈ
БЃЯеСЌНг(Safety-catch linker)ЕФЬиЕуЪЧдкФПБъНсЙЙЕФКЯГЩжаЗЧГЃЮШЖЈ. жЛгаОЬиЖЈЕФНсЙЙзЊЛЏ, ИУ linkerБЛЛюЛЏКѓВХЗЂЩњСбНтЗДгІ[23]. ЯТУцЕФЛЗыФКЯГЩвдСкмабѕБНЗгЮЊБЃЯеЪїжЌ linker НсЙЙ, дкыФСДзщзАЭъГЩКѓЭбГ§linker ЩЯЕФмаЛљ, ЪЙСкЮЛЕФЗгѕЅМќЛюЛЏ. ДЫЪБыФСД N ЖЫАБЛљКмШнвзНјЙЅЗгѕЅМќ, ЗЂЩњЗжзгФкАБНт, ЕУЕНЛЗыФВњЮя(Scheme 4)[24].
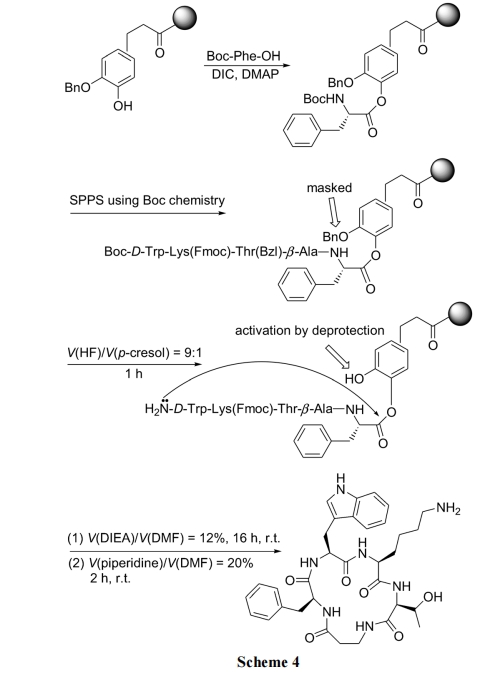
2.1.5 ЗжзгФкСђѕЅНЛЛЛ/ЗжзгФкАБНтЗЈ
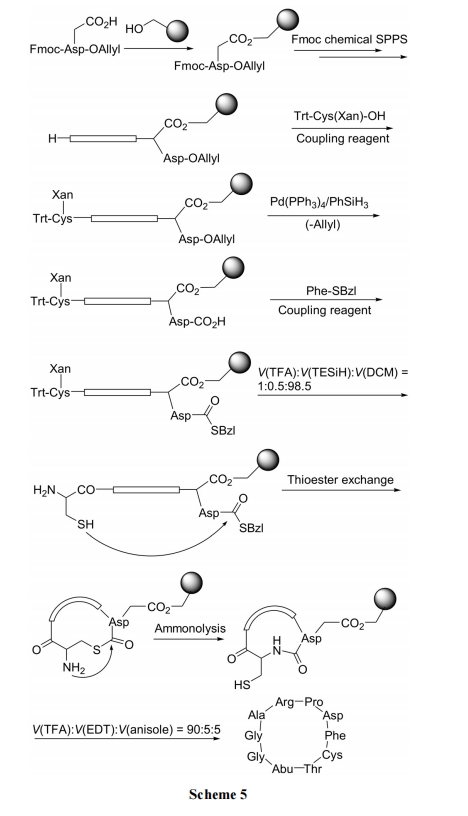
жївЊКЯГЩВНжшвРДЮЮЊ: (1)ЪЙ Fmoc-Asp-OAllyl ЕФВрСД COOH гыєЧЛљЪїжЌМќКЯЮЊмаѕЅаЭ linker; (2)ЭбГ§Fmoc, ВЂЯђ N ЖЫзщзАыФСД; (3)дк N ЖЫв§Шы Trt-Cys(Xan)зїЮЊСђѕЅНЛЛЛМААБНтЗДгІЕФЧАЬхНсЙЙ; (4)ЭбГ§ C ЖЫAsp-OAllyl ЩЯЕФ Allyl ЪЙ COOH гЮРыНјЖјгы Phe-SBzlЫѕКЯ, аЮГЩСђѕЅНЛЛЛЗДгІЕФЕзЮяНсЙЙ; (5)ЭбГ§ N ЖЫ CysЩЯЕФСНИіБЃЛЄЛљ, ЪЙ ІС-NH2МА SH гЮРы; (6)дк pH 7.5 ЬѕМўЯТНјааЗжзгФкСђѕЅНЛЛЛМААБНтЗДгІ, аЮГЩФкѕЃАЗЛЗыФ; (7) TFA ЧаГ§ Asp ВрСДЩЯЕФЙЬЯрдиЬх, ЪЙВњЮягЮРы, змЪеТЪИпДя 80% (Scheme 5)[25].
2.1.6 ЮБ Pro ажњЙбыФЛЗЛЏЗЈ
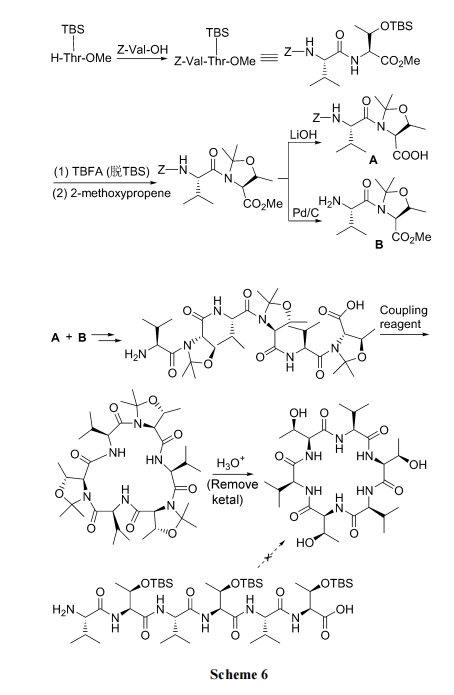
ВЛКЌ Pro Лђ Gly ЕФЖЬСДыФвђеХСІгАЯьКмФбБЛЛЗЛЏ. ЮЊДЫПЩвддкзщзАыФСДЪБАбФПБъађСажаЕФ Thr ВаЛљгУБћЭЊБЃЛЄ, днЪБЩњГЩЮхдЊЛЗ, ЪЙЦфФЃФт Pro ЕФзЊелгеЗЂНсЙЙ(turn inducer), ДяЕНМѕШѕЙбыФСДеХСІЁЂвзгкЛЗЛЏЕФФПЕФ. Д§ЛЗыФаЮГЩКѓ, ОЮТКЭЫсДІРэЭбГ§БћЭЊБЃЛЄЛљЭХ, ЛжИДThr ЕФНсЙЙ. ДЫЗНЪНЪЙВњЮязмЪеТЪгЩЃМ5%ЬсИпЕН 80%вдЩЯ(Scheme 6)[26].
2.2 ЖўСђЧХЛЗыФКЯГЩ
ДЫжжЛЗыФКЯГЩЕФЗНЗЈбЇвВКмГЩЪь. жївЊКЯГЩЗНЪНЮЊЯШзщзАыФСД, ШЛКѓЭбГ§АыызАБЫсМАЦфЫќВаЛљЩЯЕФВрСДБЃЛЄЛљ. зюКѓгУЪЪЕБЕФбѕЛЏЬѕМў, ШчПеЦј, H2O2, I2, Hg2ЃЋбЮ, Fe3ЃЋбЮ, DMSO ЕШНЋгЮРыЕФ SH бѕЛЏЮЊЖўСђМќ. гІИУзЂвтЕФЪЧдкШмвКжаНјааЗжзгФкбѕЛЏЗДгІЪББиаыПижЦгЮРыыФЕФХЈЖШВЛвЫЙ§Ип, вЛАудк 1 mmol/LжЎЯТ. ЗёдђЛсДцдкЗжзгМфДюЧХЕФИБЗДгІ. етОЭЪЧБЃжЄЗжзгФкГЩМќЕФИпЯЁЪЭддђ. ЯрБШжЎЯТ, ЙЬЯрКЯГЩЗЂЛгЕФМйЯЁЪЭаЇгІ(Pseudo-dilution effect)ПЩвдЛљБОБЃжЄЗжзгФкбѕЛЏЛЗКЯ[27]. ЕБФПБъНсЙЙжаКЌгаСНЖдвдЩЯЖўСђМќЪБ, ШчКЮе§ШЗДюЧХБуГЩЮЊЙиМќ. ЯТУцНЋНщЩмМИжжВЛЭЌЕФКЯГЩВпТд, НтОіе§ШЗаЮГЩЖўСђМќ(жїСД Folding)ЕФЮЪЬт.
2.2.1 ШШСІбЇЭЌВНКЯЛЗЗЈ
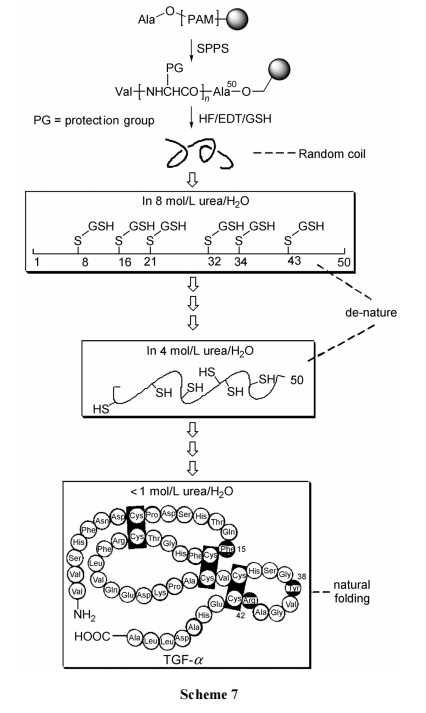
вдЙЬЯрыФКЯГЩЗНЪНКЯГЩзЊвЦЩњГЄвђзг(TGF)-ІС(КЌ50 ИіВаЛљЁЂ6 ИіАыызАБЫс)ЮЊР§. Цфжа 6 ИіАыызАБЫсЕФВрСДлЯЛљОљгУ MBzl(ЖдМзЛљма)БЃЛЄ. зюКѓвЛВНгУ HF СбНт, дкЧаГ§ЪїжЌЕФЭЌЪБвВАбАќРЈ MBzl дкФкЕФШЋВПВрСДБЃЛЄЭбГ§. гЩгкдк HF СбНтЪдМСжаМгШызуСПЕФЛЙдаЭЙШызИЪыФ(GSH), гааЇЕизшжЙСЫ TGF-ІС гЮРыыФ 6 Иі Cys ВаЛљМфЕФЫцвтМќКЯ. ШЛКѓЪЙПЊСДаЭЕФДжВњЦЗДІдкИпХЈЖШ(8 mmol/mL)ЕФыхЫиЫЎШмвКжа, ЦШЪЙ TGF-ІС ЗжзгСДЩьеЙПЊ, ГЪЁАБфадЁБ(de-nature)зДЬЌ. дйНЋДЫШмвКжУгкЭИЮіДќжа, ЫцзХДќЭтШмвКыхЫиХЈЖШж№ВННЕЕЭ, ЪЙНсКЯЬЌЕФ GSH БЛЭИЮіЧхГ§ЕФЭЌЪБ, TGF-ІС СДвВгЩдРДЕФЩьеЙЪН de-natureзДЬЌж№ВНздШЛОэЧњ. ЦфИљОнЪЧФкдДадЕФTGF-ІСОпгаЕФОэЧњзДЬЌ(гЩШ§ЖдЖўСђМќаЮГЩ)гІИУЪЧздШЛаЮГЩЕФзюЕЭФмСПзД ЬЌ . баОПвбжЄУї , TGF-ІС ЕФШ§ЖдЖўСђМќЪЧвдCys8-Cys21, Cys16-Cys32, Cys34-Cys43 ЕФаЮЪНДцдкЕФ. вђДЫ, ГЄЪБМфЁЂЛКТ§ЕиНјаае§ШЗЕФЖўСђМќДюЧХЪЧвЛИіШШСІбЇПижЦЙ§ГЬ. ШчЙћбѕЛЏЗДгІЬѕМўЙ§ЧПЁЂЪБМфЙ§Пь, ПЩФмГіЯжДэЮѓДюЧХЕФЧщПі. етжжВЛе§ШЗЕФ Folding ЛсЕМжТЩњЮяЛюадЕФЩЅЪЇ(Scheme 7)
2.2.2 е§НЛБЃЛЄ/ЗжВНКЯЛЗЗЈ
ВЩгУЖдЭбГ§ЗДгІУєИаадВЛЭЌЕФБЃЛЄЛљ, ЗжБ№БЃЛЄВЛЭЌЮЛЕу Cys ЩЯЕФВрСДлЯЛљ, ЪЙЖржж Cys дкВЛЭЌЛЏбЇЛЗОГжаЗжНзЖЮНјаабЁдёадХфЖдДюЧХ, ЪЧе§НЛБЃЛЄ/ЗжВНКЯЛЗЕФЛљБОдРэ. ДЫжжЗНЪНЕФЪЕЪЉгІзЂвтвдЯТМИЕу: (1)вђЮЊвЊОРњЖрДЮбѕЛЏГЩЛЗЗДгІ, ЮЊОЁСПМѕЩйЗжзгМфДюЧХЕФЛњЛс, гІвдЙЬЯрЛЗКЯЗНЪНЮЊвЫ. (2)ашвЊЧХСЌЕФвЛЖд Cys БиашгУЭЌжжБЃЛЄЛљ. (3)ИїЖд Cys ЕФВрСДБЃЛЄЛљБиашгыЦфЫќCys ЕФБЃЛЄЛљВЛЭЌ, ЖјЧвЫќУЧЕФЭбГ§ЗДгІУєИаадВюБ№гњДѓгњКУ, вдБуЛЅВЛИЩШХ, ЗЂЛге§НЛаЇгІ. (4)ШЋВП(ЛђГ§вЛЖд Cys вдЭт)Cys БЃЛЄЛљЕФЭбГ§ЗДгІВЛгІдьГЩЙЬЯрдиЬхЩЯ linker МќЕФЬсЧАСбНт. Scheme 8 ИјГіСЫКЌШ§ЖдЖўСђМќЕФыФКЯГЩЧщПі, ЦфжавЛЖдгУШѕЫсУєИаЕФ Trt(Ш§БНМзЛљ)БЃЛЄ; ЕкЖўЖдгУЧПЫсУєИаЕФ MoB(ЖдМзбѕЛљма)БЃЛЄ. ЫќУЧжЎМфЪєгкЫсЬнЖШВюУєИаЗНЪН. ЕкШ§ЖдгУ Acm БЃЛЄ, ЫќгыЧАСНжжБЃЛЄЛљЛЅЮЊе§НЛУєИаЗНЪН.
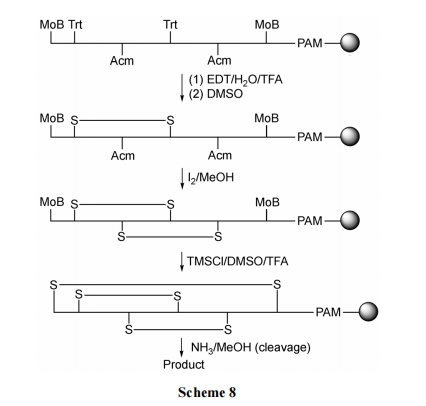
2.2.3 ЖрУзХЕСђѕЅНЛЛЛЗЈ
Tam НЬЪк[28]НЈСЂСЫвЛжжЗНБуЁЂПЩааЁЂЧЩУюЕижЦБИЖржиЖўСђЧХЛЗыФЕФКЯГЩЗНЗЈ. ДЫжжФПБъЛЗыФЕФЧАЬхБиаыдкN ЖЫОпгавЛИі Cys ВаЛљ, дк C ЖЫКЌгаСђѕЅНсЙЙ, ЖјЧвдкыФСДжаМфБиашКЌгавЛИіЛђвдЩЯЕФ Cys ВаЛљ. ШчжаМфЬх 1, дк pH ЮЊ 7.6 ЕФЫЎШмвКжа, ЪзЯШЗЂЩњСДжазюНгНќ C ЖЫЕФSHНјЙЅCЖЫСђѕЅМќЕФСђѕЅНЛЛЛЗДгІЕУЕНжаМфЬх2. ЫцКѓЗЂЩњДг C жС N ЗНЯђЕФЖрДЮСђѕЅНЛЛЛЗДгІ, ОЭЯёРСД(гжГЦЖрУзХЕ)аЇгІ, жБЕН N ЖЫзюКѓвЛИі SH ГЩЮЊСђѕЅ. ЕБШЛ, жаМфЕФCysВаЛљгжЛжИДЕНгЮРыSHзДЬЌГЩЮЊжаМфЬх3. ДЫЪБыФСДЕФ N ЖЫгы C ЖЫЯрСЌ, вђДЫ N ЖЫ Cys ЕФ NH2КмШнвзЖдзюКѓвЛИіСђѕЅНјааЗжзгФкАБНтЗДгІ, ЩњГЩФкѕЃАЗЛЗыФ 4. дк 4 жа, гЩгкЛЗЕФеХСІгАЯь, СДжаЯрСкЕФ SH КмФбаЮГЩЖўСђМќ. ЖјЮЛжУЯрЖдНЯдЖРыЕФСНИі SH жЎМфдђПЩвдаЮГЩШШСІбЇЩЯгаРћЕФЖўСђЧХ(Шч 5)ЕФНсЙЙ. зюКѓвЛЖдДјга Acm БЃЛЄЕФСНИілЯЛљдкЭбГ§ Acm Кѓ, вВГЩЮЊЖўСђЧХ, ЕУЕНЖр loop аЭЛЗыФВњЮя 6 (Scheme 9).
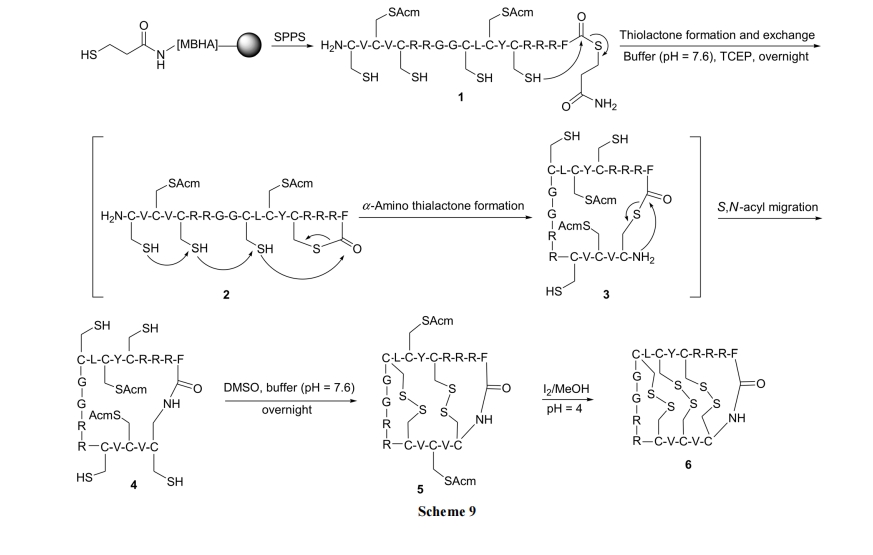
2.3 ѕЅЛЗыФ(Cyclo-depsipeptides)КЯГЩ
2.3.1 Kahalalide B ЕФКЯГЩ
дкЬьШЛВњЮяЛЗѕЅыФ Kahalalide B ЕФКЯГЩжаВЩгУСЫСНжжКЯГЩТЗЯп, ОљЕУЕНСЫФПБъВњЮя(Scheme 10)[29].
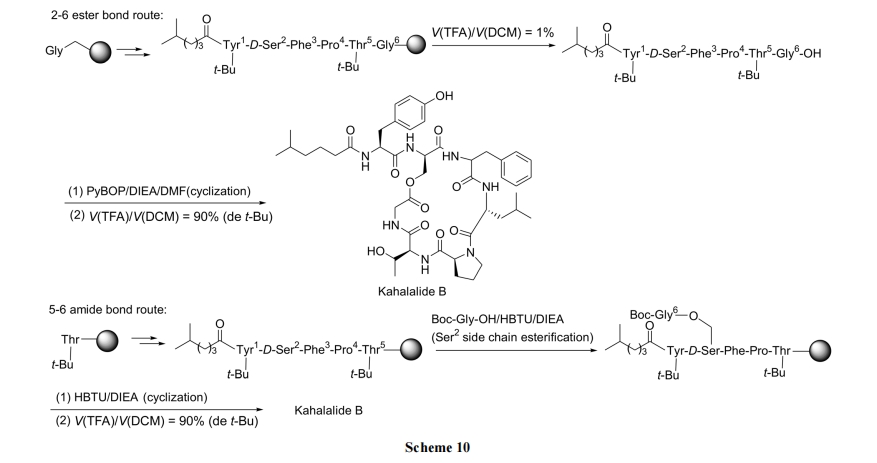
2.3.2 Kahalalide A ЕФКЯГЩ
гыЩЯР§ЕФ Kahalalide B ЯрЫЦ, БОР§ФПБъЛЏКЯЮявВЪЧКЌвЛИіФкѕЅМќЕФЛЗыФ[30]. ДгЯТУцЕФФцКЯГЩЗжЮіПЩвдЩшМЦГіЯШЙЙНЈКЌѕЅМќЕФжБСДыФ, зюКѓвдѕЃАЗМќЕФаЮЪНЙиЛЗ(Scheme 11).
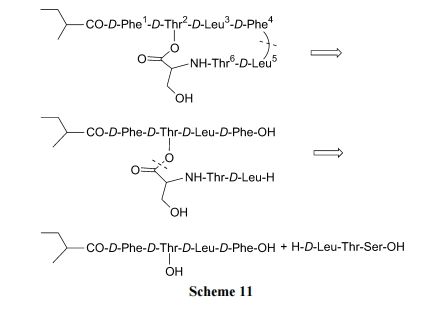
гыЩЯВЛЭЌЕФЪЧ, БОКЯГЩВЩгУЙЬЯрБЃЯеаЭ linker диЬх, ЯШНЋPhe4гыЛЧЫсаЭЪїжЌМќКЯ, ОМИВНЫѕКЯЕУЕНЮхыФжаМфЬх 7. ЭбГ§ 7 жа Thr ВрСД t-Bu КѓЪЙ OH гЮРы, вдБугы Ser7ЩњГЩѕЅМќ. ЫцКѓдйНгЩЯ Thr6МА D-Leu5, ЕУЕНжБСДаЭШЋађСаНсЙЙ 8. гЩгкЭбГ§ Fmoc ЕФЪдМССљЧтпСрЄЛсИЩШХЛюЛЏКѓЕФЛЧѕЃАЗ linker гы D-Leu5 ЕФАБЛљжЎМфЕФАБНт(зюКѓвЛВН)ЗДгІ, ЫљвдвЊдкЛюЛЏЛЧѕЃАЗМќжЎЧАЯШЪЙ D-Leu5 ЩЯЕФFmoc зЊЛЏЮЊ Trt, ГЩЮЊжаМфЬх 9. ШЛКѓгУ ICH2CN НЋЛЧѕЃАЗЕФ N дзгЭщЛљЛЏ, ЪЙБЃЯе linker ЛюЛЏ. дйгУ TFA ЭбГ§N ЖЫЕФ Trt, ЕУЛЗЛЏЧАЬх 10. зюКѓдк DIEA зїгУЯТЗЂЩњЗжзгФкАБНт , ЭЌ ЪБ Ча Г§ Ъї жЌ ЕУЕНВњЮя Kahalalide A (Scheme 12).
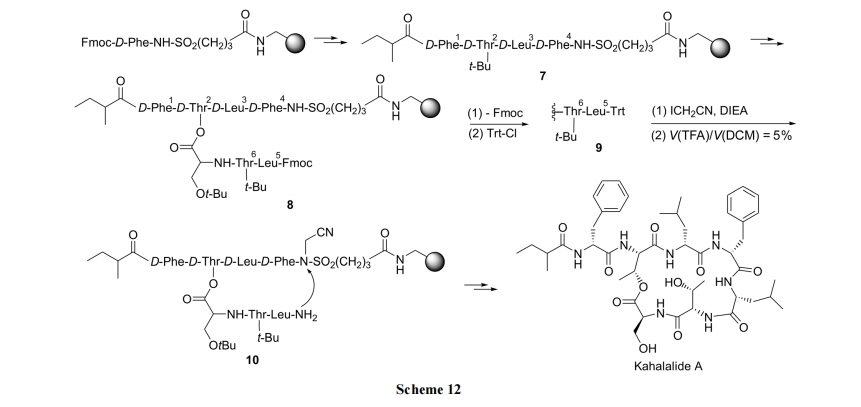
2.3.3 здЖЏгеЕМыФ AIP ЕФКЯГЩ
БОР§КЯГЩЕФФПБъЛЏКЯЮя AIP (auto-inducing peptide, здЖЏгеЕМыФ)ЮЊВр-ЮВвдѕЅМќГЩЛЗЕФЛЏКЯЮя. ИУКЯГЩВпТдЕФЬиЕуЪЧ: (1)вдѕЃыТЮЊЬиЪтСбНтаЭ linker; (2)дк linker ЩЯЪзЯШМќКЯЕФВЛЪЧыФСД C ФЉЖЫЕФЕквЛИіВаЛљ, ЖјЪЧЕкЖўИіВаЛљ; (3) C ЖЫЕквЛИіВаЛљгыыФСДжаМф Ser ВаЛљЕФВрСДOH вдѕЅМќЯрСЌ. ЩшМЦДЫжжКЯГЩЗНЪНЕФвРОндкгкѕЃЛЏБНыТ linker ЖдгкыФКЯГЩжаЕФЗДгІЬѕМўФЭЪмадКмЧП, ЩѕжСдкЫсЁЂМюЛЗОГжавВКмЮШЖЈ. ШЛЖјДЫжж linker НсЙЙЖдбѕЛЏЬѕМў, Шч Cu2ЃЋ, I2, NBS ЕШЗЧГЃУєИа, МЋвзЪмЕНЧзКЫЛљЭХЕФНјЙЅЖјСбНт. вђДЫ, Ser ВрСДЩЯВаЛљЕФ NH2ПЩвдЗНБуЕиЖдѕЃыТ linker НјааЗжзгФк SN2 ЗДгІ, аЮГЩѕЃАЗМќ, ЭЌЪБЧаГ§ЪїжЌ(Scheme 13)[31].
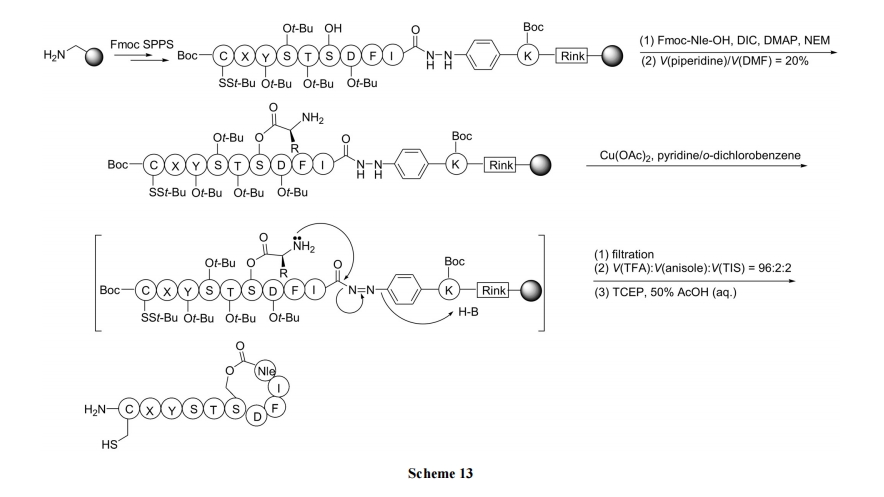
2.3.4 fпђЭЊѕЅНЛЛЛЗЈ
Heimgartner ЕШ[32]ЩшМЦСЫвЛжжУћЮЊЁАжБНгѕЃАЗЛЗЛЏ(direct amide cyclization)ЁБЕФКЯГЩТЗЯп, жЦБИСЫгЩ ІС,ІС-ЫЋШЁДњВаЛљзщГЩЕФѕЅЛЗыФ. ЦфдРэЪЧжБСДыФ C ЖЫЕФЖўМзЛљѕЃАЗНсЙЙдкЮо ЫЎ HCl Дп ЛЏ ЯТ , ЯШЩњГЩвЛИіЁАazirine/oxazoloneЁБЙ§ЖЩЬЌ 11 МА 12. КѓепЪмЕНЭЌЗжзгСДN ЖЫєЧЛљЕФНјЙЅ, ЪЙ oxazolone ЮхдЊЛЗНтЬх, ЭЌЪБгы OHаЮГЩЗжзгФкѕЅМќ(Scheme 14).
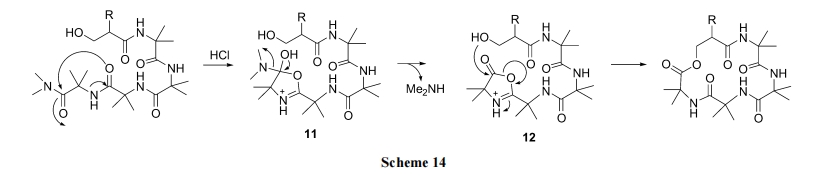
ДгжаПЩвдПДГі, ІС,ІС-ЫЋШЁДњАБЛљЫсЕФНщШыЪЧЗЧГЃживЊЕФ, ЫќУЧШЗБЃСЫыФСД N ЖЫєЧЛљЯђ C ЖЫЕФНгНќ, РћгкѕЅЛЗЕФаЮГЩ. Koch ЕШ[33]РћгУДЫЗНЪНКЯГЩСЫвЛаЉѕЅЛЗыФ 19, ЛЗЛЏВњТЪДя 46% (Scheme 15).
2.3.5 Ugi ЗДгІбмЩњЗЈ
вдШЉЁЂввыцєШЫсѕЅЁЂАЗШ§жжзщЗж(ЫФжжЙІФмЛљ)ЮЊЕзЮяНјааЕФ Ugi ЗДгІ, вВПЩвдЕУЕНКЌвЛИіѕЅМќЕФѕЅЛЗыФВњЮя. Цфжа, ЯШгЩ CHO, NC, NHR Ш§жжЙІФмЛљЫѕКЯ, ЩњГЩfпђжаМфЬх 20. дйОЗжзгФкєШЫсѕЅгеЗЂЕФжиХХ, ОТнЭщаЭЫЋfпђжаМфЬх 21, зюКѓЩњГЩЛЗѕЅМќ(Scheme 16) [34].
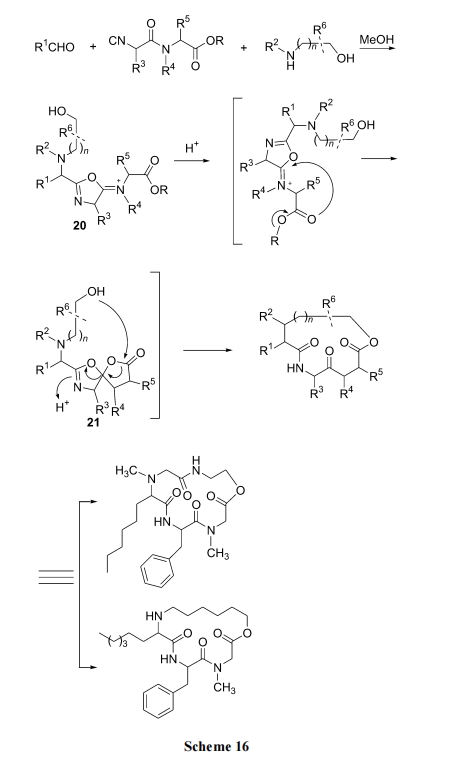
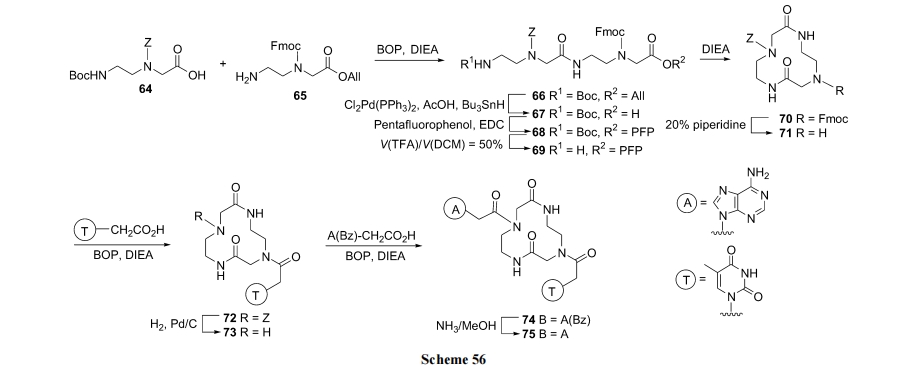
3 DKP ЛЗыФбмЩњЮяКЯГЩ
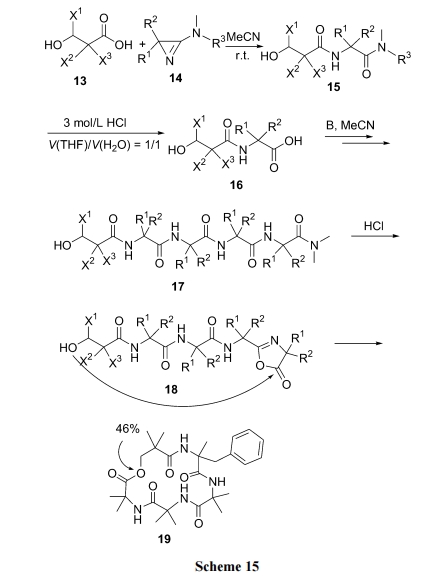
3.1 ЖўыФѕЅЕФЗжзгФкАБНт
ЮоТлЪЧОљЯрШмвКЗЈжЦБИЕФЖўыФѕЅ, ЛЙЪЧвдѕЅМќЮЊlinker ЕФЙЬЯрдиЬхЩЯЕФЖўыФ, ЕБ N ЖЫЕФБЃЛЄЛљБЛЭбГ§Кѓ, дкШѕМюЛђ HOAc ЕФДпЛЏЯТКмШнвзЗЂЩњгЮРы NH2 ЖдЗжзгФкЕФ C ФЉЖЫѕЅМќЕФАБНтЗДгІ(Scheme 17).
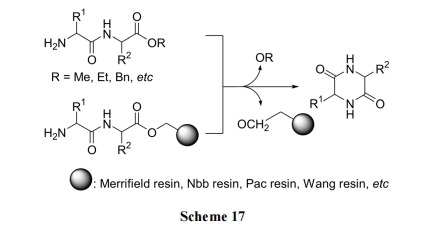
жЕЕУжИГіЕФЪЧЩЯЪіЗДгІдкОЕфЕФвдмаѕЅМќЮЊ linkerЕФЙЬЯрыФКЯГЩжаЪЧвЛИіПЩЕМжТ(Ш§ыФвдЩЯЕФ)ШЋКЯГЩЪЇАмЕФбЯжиИБЗДгІ. ЮвУЧ[35]е§ЪЧРћгУетИіИБЗДгІ, вд Pac ЪїжЌЮЊдиЬхжЦБИСЫЖржжЪеТЪРэЯыЕФ DKP ЛЗЖўыФВњЮя(Scheme 18)
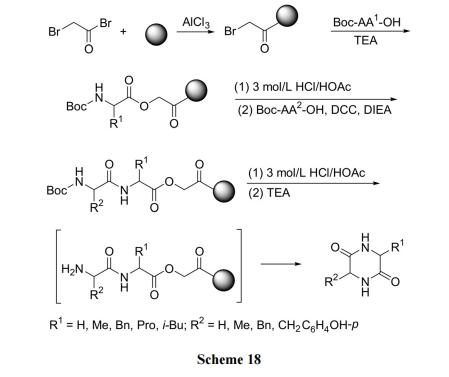
3.2 BAL ЪїжЌЮЊдиЬхЕФЖўыФЗжзгФкЛЗЛЏ
ЙЬЯрЩњГЩ DKP ЕФЗНЪНвЛАуЛљгк DKP ЧАЬхЕФєШЛљгыЪїжЌЕФ linker вдѕЅМќаЮЪНжБНгЯрСЌ, вђДЫАБЛљНјЙЅѕЅМќЗЂЩњЗжзгФкЛЗЛЏЪБаЮГЩ DKP ВЂЭЌЪБДгЪїжЌЩЯЧаИю. Жјвд BAL ЪїжЌЮЊдиЬхЕФ DKP КЯГЩЪЧЭЈЙ§ linker гы DKPЧАЬхЕФАБЛљЯрСЌ, ЫљвдЪЧЯШаЮГЩ DKP, КѓНјааЪїжЌЧаИю, МДСНВНЗДгІЗжПЊНјаа. етбљ, дкЩњГЩ DKP Кѓ, ЭЈЙ§ЯДЕгГ§ШЅШмвКжаЕФдгжЪ, ЧаИюКѓЕУЕНИпДПЖШЕФВњЮя[36](Scheme 19).
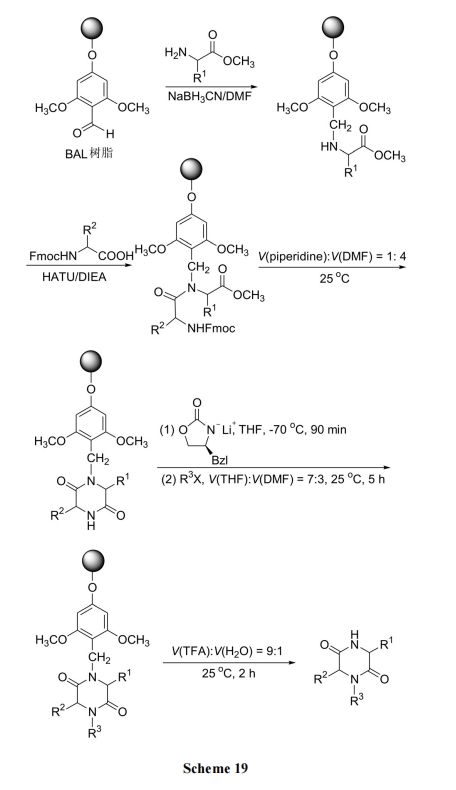
3.3 ШЉгыАБЛљЫсЕФЛЙдЭщЛљЛЏЭООЖКЯГЩ DKP бмЩњЮя
СЌдк Wang ЪїжЌЩЯЕФЕквЛИіАБЛљЫсЕФ NH2гыЯргІЕФШЉЯШЫѕКЯГЩ Schiff Мю, КѓепдкЛЙдМС Na(OAc)3BH зїгУЯТ, ЩњГЩNЭщЛљЛЏжаМфЬх22. ЫцКѓгУСэвЛИіBoc-АБЛљЫсЖд 22 ЕФжйАЗНјааѕЃЛЏЕУ 23. ЭбГ§КѓепЕФ Boc КѓдйгкМзБНжаМгШШ, ЗЂЩњЗжзгФкАБНтВЂЭЌЪБЧаГ§ЪїжЌ, ЕУЕН N ЩЯгаШЁДњЕФ DKP ВњЮя[37] (Scheme 20). Г§СЫЩЯУцЕФ Na(OAc)3BH Эт, ЛЙПЩвдЪЙгУ NaCNBH3 ЮЊЛЙдМС, ЙЬЯрдиЬхПЩгУ PAM ЪїжЌДњЬцНЯАКЙѓЕФWang ЪїжЌ, ЕУЕНСЫЪеТЪИќТњвтЕФВњЮя[38].
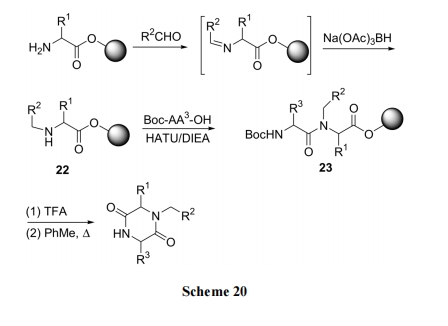
3.4 N,N'-ЫЋШЁДњ DKP бмЩњЮяЕФКЯГЩ
гУ 2-ЯѕЛљБНЛЧѕЃТШЖдЪїжЌЩЯЕквЛИіАБЛљЫсЕФ NH2Нј аа Сй ЪББЃЛЄ , ЪЃ ЯТвЛИі H д зг ШЗ БЃ дк Ыц Кѓ ЕФMitsunobu ЗДгІЕУЕН N ЕЅШЁДњВњЮя 24. ЭбГ§ N ЩЯЛЧѕЃЛљКѓдйБЛфхввѕЃЛЏ, ЕУжаМфЬх25. ОSN1ЗДгІНЋ25зЊЛЏЮЊN ШЁДњЕФИЪАБѕЃЖўыФ 26. Кѓепдк TFA-H2O ДпЛЏЯТЩњГЩжБСДЖўыФ 27 МА DKP ЛЗЖўыФ 28 ЕФЛьКЯЮя. дкШ§ЗњввЫсєћ(TFAA)/DCMМАЮТШШЯТ, 27ПЩЭъШЋзЊЛЏЮЊФПБъВњЮя28[39](Scheme 21).
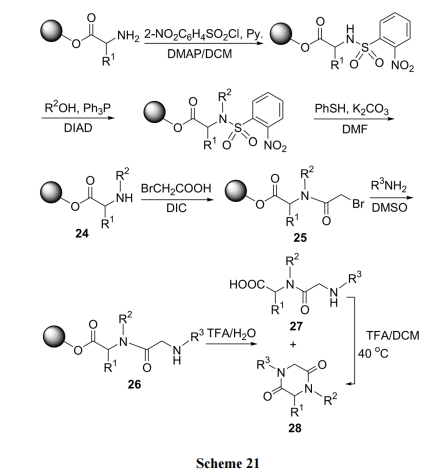
3.5 Ugi ЗДгІЭООЖКЯГЩ DKP бмЩњЮя
Ugi ЗДгІЪЧгУШЉЁЂЫсЁЂАЗМАвьыц 4 жжЗДгІЕзЮяНјааЖрзщЗжвЛЙјЗДгІ(MCR). гЩгкЗДгІЕзЮяРраЭЖр, вђДЫВњЮяОпгаКмКУЕФНсЙЙЖрбљад. гШЦфЪЧЙЬЯрЗДгІЗНЪН, ПЩвдМђЛЏЗДгІКѓЕФДПЛЏЙ§ГЬ, МДГ§СЫЙвдкЙЬЯрдиЬхЩЯЕФЫѕКЯВњЮяЭт, ЦфЫќЪЃгрЕФМИжжЗДгІЮяОљПЩЙ§ТЫГ§ШЅ[40](Scheme 22).
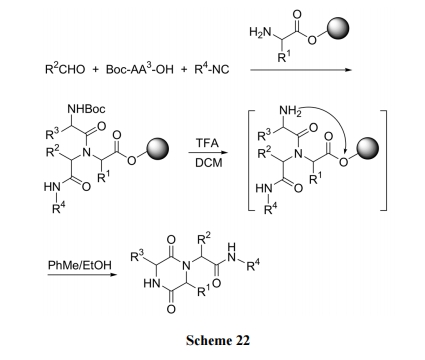
3.6 ВЂЛЗ DKP бмЩњЮяЕФКЯГЩ
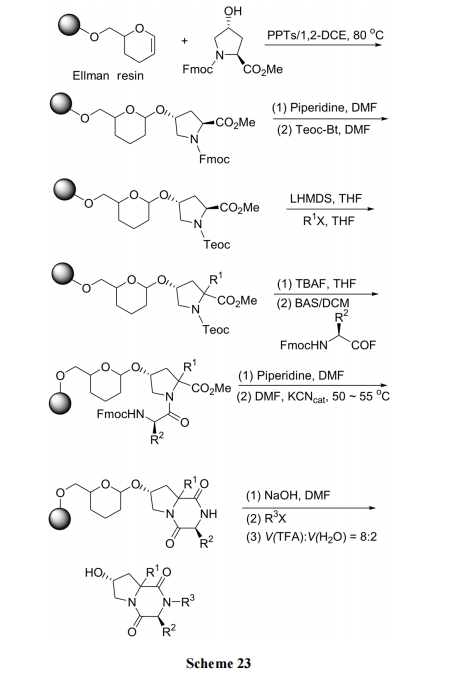
аэЖрИпЛюадЕФ DKP аЭЬьШЛВњЮяМАШЫЙЄЩшМЦКЯГЩЕФDKP бмЩњЮяЕФНсЙЙдЖБШЩЯЪіКЯГЩР§ЕФВњЮявЊИДдгЕУЖр, ЫќУЧЭљЭљОпгаВЂЛЗЕФЙЧМмНсЙЙ. ДЫРрЛЏКЯЮяЕФКЯГЩвбгавЛаЉЮФЯзБЈЕР[41,42]. ЯТУцЕФКЯГЩЪЧИЌАБЫсЕФЮхдЊЛЗЮЊжЦБИ DKP ВЂЛЗЧАЕФжЇМмНсЙЙ. ЪзЯШНЋСНЖЫБЃЛЄЕФєЧИЌАБЫс Fmoc-Hyp-OMe ЕФВрСД OH гы Ellman ЪїжЌЕФЫЋМќЗЂЩњМгГЩЗДгІЖјМќСЌЩЯ. ЫцКѓЪЙ Hyp ВаЛљЩЯ ІС-ЬМЭщЛљЛЏ, дйгУЕкЖўжжАБЛљЫсгы Hyp НгГЩЖўыФ. ЭбГ§ЕкЖўЮЛВаЛљЕФN БЃЛЄКѓ, ЪЙгЮРы NH2НјЙЅ Hyp ЕФМзѕЅМќ, ЗЂЩњЗжзгФкАБНт, ЩњГЩ DKP ВЂЛЗНсЙЙ(Scheme 23) [42].
3.7 ЯЉВрСД DKP ЕФКЯГЩ
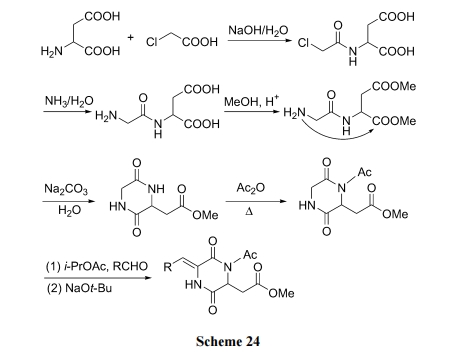
ЪзЯШгУСЎМлЕФТШввЫсгыгЮРыЕФЬьЖЌАБЫсЫѕКЯ, дйгУАБЫЎгыТШЗЂЩњ SN2 ЗДгІжЦЕУ Gly-Asp ЖўыФ. КѓепдкЫсадМзДМЛЗОГжаЪЙ Asp ЕФєШЛљМзѕЃЛЏ, зюКѓОЗжзгФкАБНтЩњГЩ DKP аЭВњЮя. ШЋВПКЯГЩЕФзюДѓЬиЕуЪЧВЛгУОЕфЕФыФЫѕКЯЪдМС, вВУЛгаШЮКЮБЃЛЄгыЭбБЃЛЄЕФЗДгІ, вђДЫГЩБОЕЭЁЂЪЪгкДѓЩњВњ[43] (Scheme 24).
3.8 ыФЫЋЗжзгЫѕКЯЗЈ
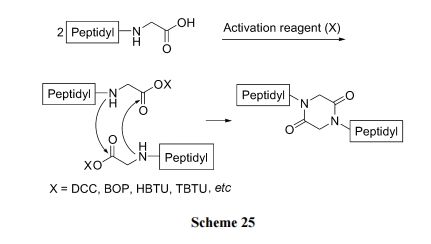
ЧАУцНщЩмЕФИїжжКЯГЩжа, вЛАуОљвЊОРњЖўыФЗжзгФкАБНтаЮГЩ DKP ЛЗЭООЖ. ЦфЪЕыФзїЮЊЕЅЬхЕФЫЋЗжзгЫѕКЯвВЪЧвЛжжКЯГЩ DKP НсЙЙЕФКЯРэЗНЪН. вђЮЊетжжЗНЪНЕУЕНЕФ DKP бмЩњЮяОпга N,N'-ЫЋШЁДњЕФНсЙЙЬиЕу. ДЫжжКЯГЩашвЊЗДгІЮяЕФ C ФЉЖЫКЌга Gly ВаЛљ, вђЮЊЦфЫќВаЛљЕФІС-ЬМдзгЩЯОљгаВрСДШЁДњ, ЮЛзшМгДѓВЛвзЗЂЩњЫЋЗжзгЗДгІ. баОПЗЂЯж, аэЖрЙбыФОЫЋЗжзгЫѕКЯаЮГЩЕФDKPЫЋБЖыФСДНсЙЙКѓ, ЦфдгаЕФЛюадПЩЛёЕУЬсИп. вђДЫКЌDKPЛЗЕФ ЫЋ Вр ыФЛЏКЯЮяЕФ Щш МЦ гыКЯГЩОпгавЛЖЈ вт вх [44](Scheme 25).
3.9 ТнЛЗ DKP ДЎСЌыФЕФКЯГЩ
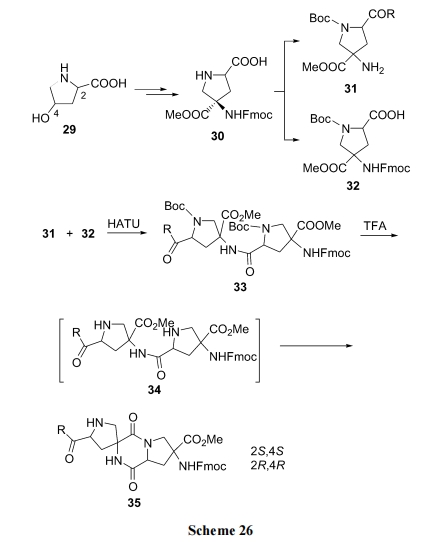
вдєЧИЌАБЫс(29)ЮЊдСЯ, ОЖрВНзЊЛЏЩњГЩЙиМќжаМфЬх 30, дйНЋ 30 ЗжБ№зЊЛЛЮЊАБЛљзщЗж 31 МАєШЛљзщЗж 32. СНИізщЗжЫѕКЯЮЊЖўыФ 33 жЎКѓ, ЭбГ§гЩ 32 бмЩњЕФНсЙЙЖЮЩЯЕФ Boc, ЪЙжйАБЛљгЮРыЕУ DKP ЧАЬхНсЙЙ 34. ЫцКѓдкМюЛђЫсДпЛЏЯТ, ЗЂЩњЖўыФЗжзгФкАБНт, ЩњГЩвЛжжЮхдЊЛЗгыDKP СЌЮЊТнЛЗЕФВњЮя 35. етОЭЪЧТнЛЗ DKP ДЎСЌыФжаЕФвЛИіЕЅдЊНсЙЙ[45] (Scheme 26).
4 КЌУбЧХЛЗыФКЯГЩ
ДгЧХСЌНсЙЙПД, УбЛЗыФгжЗжЮЊжЌ-ЗМЛьУбаЭМАЗМУбаЭСНДѓРр, ВЂвдЧАепЮЊЖрЪ§. ИљОнУбЧХЕФЩњГЩЗНЪН, гаScheme 27 ЫљЪОЗДгІЭООЖ.
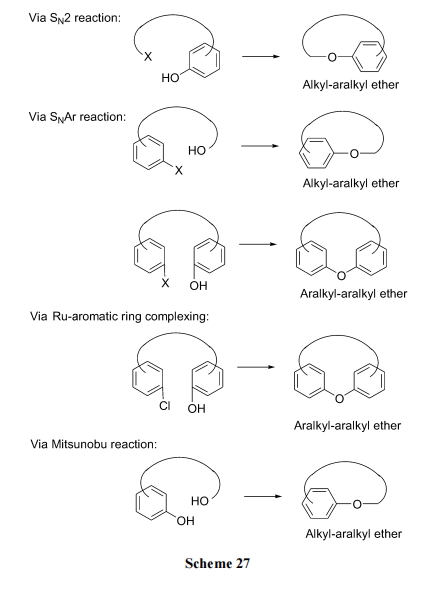
дкКЯГЩВпТдЩЯгыѕЅЛЗыФЯрЫЦ, УбЛЗыФЕФКЯГЩЫГађЩЯвВДцдкСНжжЧщПі: (1)ЯШжЦБИыФСДжїЬхНсЙЙ, зюКѓвдУбМќЕФЩњГЩЪЕЯжКЯЛЗ. (2)ЯШжЦБИКЌУбНсЙЙЕФЦЌЖЮ, зюКѓвдѕЃАЗМќ(ЛђЦфЫќМќ)ЕФЩњГЩЪЕЯжКЯЛЗ.
4.1 ЛьУбЛЗыФКЯГЩ
ЬьШЛВњЮяУбЛЗыФ Sanjoinine G1 ЕФЙЧМмЩЯКЌгаСНИіыФМќМАвЛИіЗМЬў/ЭщЬўЛьКЯУбМќ. дкФцКЯГЩЗжЮіетИіЪЎЫФдЊЛЗЕФФПБъНсЙЙЪБ, ПЩвдШЗЖЈСНИіГЩЛЗМќЮЛжУ a гы b (Scheme 28). вђДЫДцдкСНЬѕТЗЯпПЩвдЭъГЩДЫЛЗыФЕФКЯГЩ.
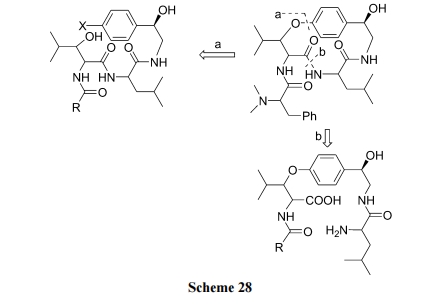
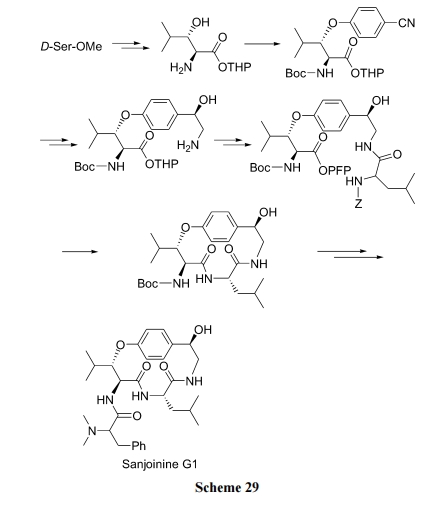
ТЗЯп(a), ЯШжЦБИКЌгаЛьУбНсЙЙЕФжБСДыФ, зюКѓНјааѕЃАЗМќЗНЪНЙиЛЗ(Scheme 29)[46].
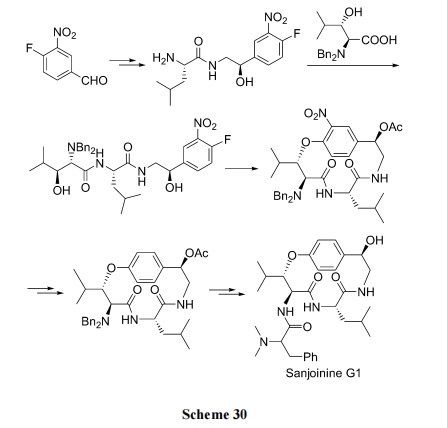
ТЗЯп(b), ЯШжЦБИыФСД, зюКѓО SNAr ЗДгІЪЙЗњДњБНгыВрСДЩЯЕФжйДМжЎМфЭбвЛЗжзг HF ЩњГЩЛьУбМќ(Scheme 30)
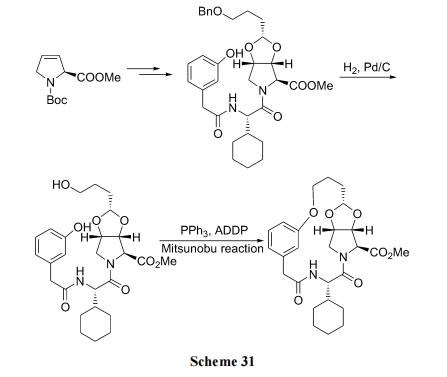
РћгУ Mitsunobu ЗДгІжЦБИ ЛьУбЛЗФтыФ(Scheme 31)[47].
РћгУыФСДжа Tyr ЕФВрСДЗгєЧЛљгыЗжзгФкСэвЛЮЛЕуВрСДЩЯЕФфхдзгжЎМфЕФSN2ЗДгІЩњГЩЛьУбЛЗыФЪЧБШНЯМђБуЕФКЯГЩЗНЪН(Scheme 32)[48].
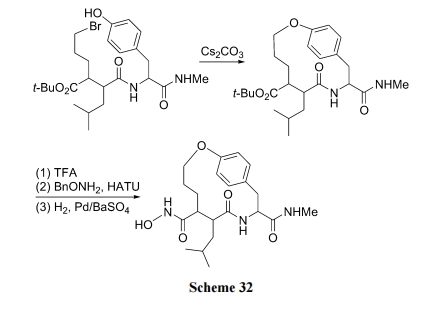
ЭѕЕТаФЕШ[49]вдыФСД N ЖЫЕФфхввѕЃМА C ЖЫЕФ Tyr ЮЊСНИіЧХЭЗ, ОЙЬЯрыФКЯГЩЗНЪНЪзЯШжЦБИКЌетСНжжЧХЭЗНсЙЙЕФжБСДыФжаМфЬх, зюКѓвд K2CO3ЮЊМюДпЛЏМСЗНБуЕиЙЙНЈСЫвЛаЉЛьУбЛЗыФВњЮя(Scheme 33).
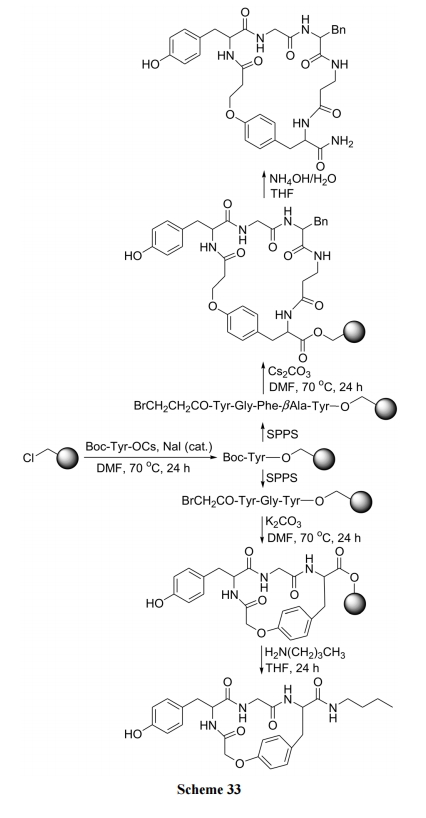
гыЩЯР§КЯГЩВЛЭЌЕФЪЧНЋТБдзгСЌдкБНЛЗЩЯ, єЧЛљЮЛгкЭЌЗжзгЕФЭщЛљСДЩЯ. гЩгкЖдЮЛЯѕЛљЕФгАЯь, F дзггаНЯИпЕФЗДгІЛюад, гУ K2CO3ЕФМюадЬѕМўМДПЩЭъГЩЛьУбМќЛЗКЯ(Scheme 34)[50].
4.2 ЗМУбЛЗыФКЯГЩ
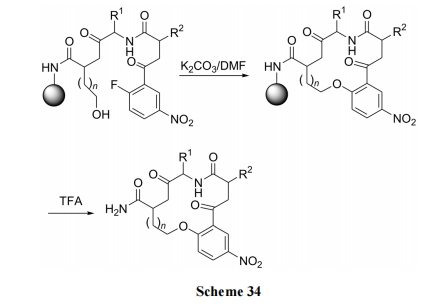
ЪзЯШвдОпгаСкЯѕЛљЗњБНШЁДњЕФвьыцМАЦфЫќЕФШ§жжзщЗжНјаа 4CR (four-component reaction)ЗНЪНЕФ Ugi ЫѕКЯЗДгІ, жЦЕУКЌЗгєЧЛљМАСкЯѕЛљЗњБНЕФжБСДыФжаМфЬх. ЫцКѓдкМюДпЛЏЯТЗЂЩњЗжзгФк SNAr ЗДгІ, ЕУЕНЗМУбЛЗыФВњЮя(Scheme 35)[51].
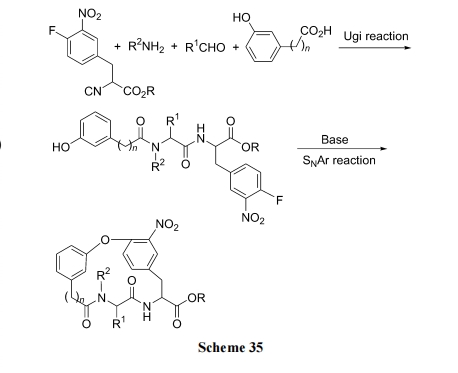
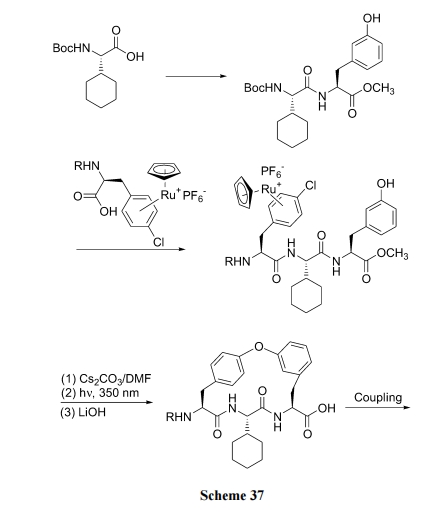
Г§СЫЩЯР§КЯГЩжав§ШыСкЁЂЖдЮЛ NO2Лђ CN ПЩвдЛюЛЏЗМТБдзгЕФЛюадЭт, юЩЪдМСгыТШБНаЮГЩ ІЧ6-ЗМЯЉТчКЯЮяЙ§ЖЩЬЌЕФЗНЪНЬсИпБНЛЗЩЯТШдзгЕФЗДгІЛюад. ОпЬхТЗЯпЪЧЯШЪЙ Boc-Phe(4-Cl)OH гыюЩЪдМСаЮГЩЛюЛЏТчКЯЮя 36, ШЛКѓдкєШЛљЗНЯђзщзАыФНсЙЙЕУжаМфЬх 37, Кѓепдк Na ЪдМСДпЛЏЯТ, Г§ШЅ HCl ЩњГЩЗМУбМќ. зюКѓОЙтНтЗДгІГ§ШЅТчКЯЕФюЩбЮ, ЕУФПБъВњЮя(Scheme 36)[52].
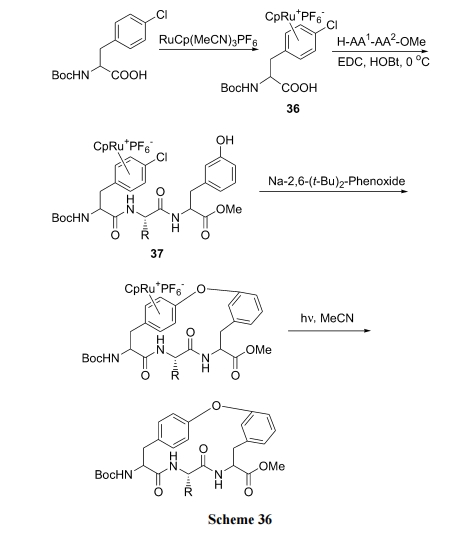
ЛЙПЩгУ Cs2CO3/DMF гыюЩЪдМС-ТШБНТчКЯЮяЗДгІЭъГЩДЫВНЛЗКЯ(Scheme 37)[53].
5 ЕЅСђМАЖрСђУбЛЗыФКЯГЩ
Г§СЫЧАУцНщЩмЕФОЕфЖўСђЧХвдЭт, ЪєгкСђУбЧХНсЙЙЕФЛЙгаЕЅСђУбЧХЁЊSЁЊЁЂбЧМзЖўСђУбЧХЁЊSCH2SЁЊМАСЌШ§СђЧХЁЊSЁЊSЁЊSЁЊЕШаЮЪН. ЦфжавдЕЅСђЧХНЯЮЊЖрМћ, ЫќУЧгжЗжЮЊЭщЬўЕЅСђУбМАЗМЭщЛьСђУбСНжж. вђИїжжСђУбЧХЕФНсЙЙВЛЭЌ, жЦБИЗНЗЈПЩЗжЮЊвдЯТМИжж: (1)КЌЕЅСђУбЦЌЖЮЕФЦфЫќМќ(вд CONH ЮЊжї)ЕФЛЗКЯ; (2) SN2 ЗДгІЗНЪН, вдыФСДжа Cys ВаЛљВрСДЕФлЯЛљЮЊЁЊSЁЊЕФЧАЬхНсЙЙ, вдЭЌЗжзгФкЕФТБдзгЮЊЧзКЫЛљЭХ; (3) SNAr ЗДгІЗНЪН, вдCys ВрСДЕФлЯЛљМАЭЌЗжзгФкЗМЛЗЩЯЕФЗњдзгЮЊЛЗКЯЗДгІЕФЧХЭЗзщЗж.
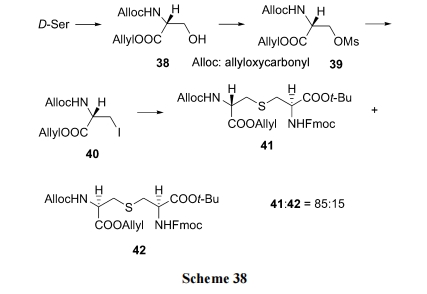
5.1 дЄжЦЕЅСђУбжаМфЬхЕФЛЗыФКЯГЩ
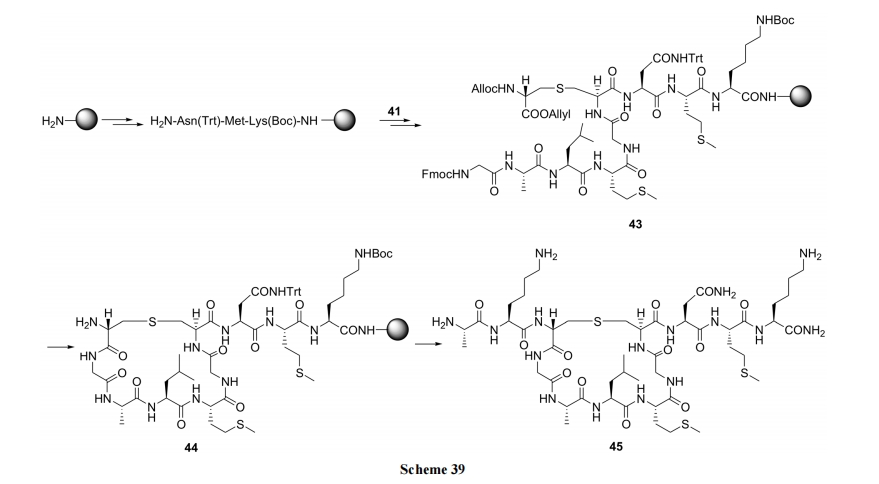
Tabar ЕШ[54]НЈСЂСЫвЛЬѕПЩвджЦБИВњЮяЕФСЂЬхбЁдёадНЯИпЕФКЯГЩТЗЯп. вд D-ЫПАБЫсЮЊдСЯ, ЯШКѓОСНВрБЃЛЄЁЂєЧЛљМзЛЧЫсѕЅЛЏЁЂЕтШЁДњЕУЕНживЊжаМфЬхВрСДЕтДњБћАБЫс(40). КѓепдкМюДцдкЯТ, гыЫЋБЃЛЄЕФАыызАБЫсЗЂЩњ SN2 ЗДгІ, ЕУЕНвЛЖдВњТЪВюБ№УїЯдЕФвдЕЅСђУбЮЊЧХЕФЫЋБћАБЫсбмЩњЮя Lanthionine (41). КѓепзїЮЊвЛИіЙиМќЙЙМўЭљЭљгУгквЛРрУћЮЊ Lantibiotics ЕФЬьШЛВњЮяыФКЯГЩ(Scheme 38). Tabar ЪЕбщЪвдђНЋ 41 гУгкЬьШЛВњЮя Nisin CЛЗРрЫЦЮя 45 ЕФЙЬЯрКЯГЩ(Scheme 39)[55].
5.2 вд SN2 ЗДгІЗНЪНЙиЛЗжЦБИЕЅСђУбЛЗыФ
вдSN2ЗДгІЗНЪНЙиЛЗЪЧжЦБИЕЅСђУбЛЗыФзюГЃгУЕФВпТд. ЦфжаЬсЙЉСђдзгЕФНсЙЙЖрЮЊыФСДжаЕФ Cys ВаЛљ, ЖјКЌТБдзгЕФНсЙЙЮЊВрСДбмЩњЕФфхЭщСДЛђ N ЖЫфхввѕЃ(ЛђТШввѕЃ). МюДпЛЏЪЧБиашЕФЗДгІЬѕМў, вЛАуЕФЪхАЗ[Ш§ввАЗЁЂN-МзЛљТ№пјЛђ DBU (1,8-diazabicyclo[5.4.0]-undec-7- ene)]зувдБЃжЄ SN2 ЗДгІЕФНјаа. DBU ДпЛЏЕФЙиЛЗМћ Eq. 1[56]; Ш§ввАЗ/ЫЎДпЛЏЕФЙиЛЗМћ Eq. 2[57].
5.3 дкыФСД N ЖЫв§ШыЖдЮЛЯѕЛљЗњДњБН
гЩгкЯѕЛљЕФРЕчзгаЇгІ, ЪЙЖдЮЛ F дзгЛюЛЏ, зувдгы SH ЗЂЩњ SNAr ЗДгІ, ЩњГЩЗМЭщЛьСђУбМќ(Scheme 40)[58].
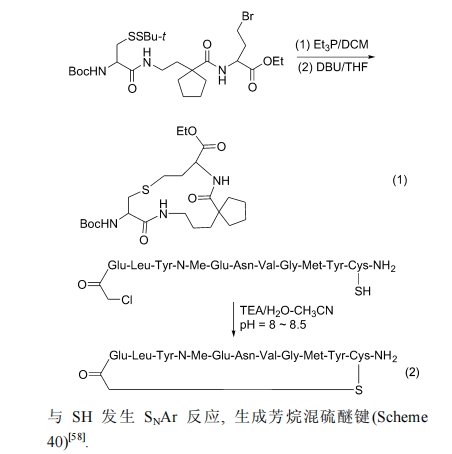
5.4 Michael МгГЩЗЈ
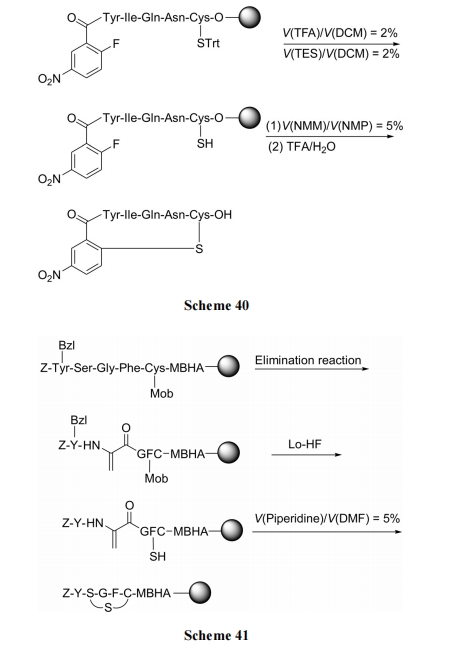
дкыФСДжаЪЪЕБЮЛЕуЕФ Ser ВаЛљЩЯНјааЯћГ§ЗДгІ, ЩњГЩЭбЧтБћАБЫс(Dha), ШЛКѓгыыФСДЩЯЕФгЮРы SH ЗЂЩњMichael МгГЩЕУЕНЕЅСђУбЛЗыФ, зюКѓдйЭбГ§ВрСДБЃЛЄМАЧаГ§диЬх. ДЫжжЗНЪНгжБЛШЯЮЊЪЧФЃФтЩњЮяКЯГЩЕФвЛжжЭООЖ(Scheme 41)[59].
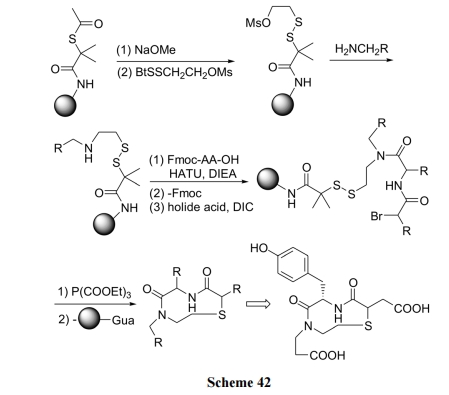
5.5 РћгУ N ЖЫЕФфхввЫсЖдЖўСђМќаЭ linker НјааЛЗЛЏСбНт
БОР§КЯГЩЕФЬиЕуЪЧРћгУ N ЖЫЕФфхввЫсЖдЖўСђМќаЭlinker НјааЛЗЛЏСбНтЗДгІ. ВњЮяОпгаЕфаЭЕФ ІТ-turn ФтыФНсЙЙ(Scheme 42)[60].
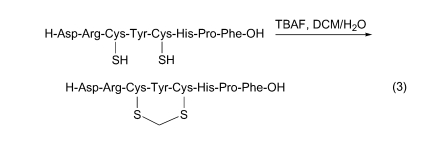
5.6 ЩњГЩбЧМзЖўСђУб(SCH2S)Мќ
ыФСДжаКЌСНИілЯЛљЪЧжЦБИСЌЖўСђМќЛЗыФЕФНсЙЙЛљДЁ. ЕЋШчЙћдкЯрзЊвЦДпЛЏМСЫФЖЁЛљЗњЛЏяЇДцдкЯТ, ПЩвдЛюЛЏШмМС CH2Cl2, ЪЙКѓепЩњГЩПЈБіВЂНщгкСНИіСђдзгжЎМф, ЩњГЩбЧМзЖўСђУб(SCH2S)Мќ(Eq. 3)[61].
5.7 СЌШ§СђЧХЛЗыФЕФКЯГЩ
СЌШ§СђЧХЛЗыФДцдквЛаЉЬьШЛВњЮяжа, ЫќУЧОпгаЕФПЙОњМАЯИАћЖОЛюадвбОв§Ц№ЛЏбЇМвЕФЙизЂ. жЦБИетжжЛЗыФЕФЛљБОдРэЪЧвдКЌгаСНИі Cys ВаЛљЕФыФЮЊЕзЮяНсЙЙ, ЦфжаЕФвЛИіCysЩЯЕФлЯЛљЯШгызЈУХЕФСђУбЛЏЪдМСЩњГЩЛьСЌЖўСђМќЛђЛьСЌШ§СђМќ. ШЛКѓдйНгЪмЗжзгФкСэвЛИілЯЛљЕФНјЙЅ, ЗЂЩњСђНЛЛЛЗДгІ, ЩњГЩСЌШ§СђМќЛЗыФ. вд TBPI (N,N'-СђЫЋСкБНЖўМзѕЃбЧАЗ)ЮЊСђУбНЛЛЛМСМћ Scheme 43[62]. вд Mpa(МзбѕѕЃСЌШ§СђБћЫс)ЮЊСђУбНЛЛЛМС, Мћ Scheme 44[63].


5.8 ЖрСЌСђУбЛЗыФЕФКЯГЩ
НќФъЗЂЯжвЛаЉКЌга5ИіЩѕжСИќЖрЕФСЌСђУбЕФЬьШЛВњЮя, вђДЫЛЏбЇМвПЊЪМЖдЖрСЌСђУбЛЗыФЕФКЯГЩНјааСЫбаОП. БШНЯгаДњБэадЕФвЛжжЗНЗЈОЭЪЧЯШЙЙНЈКЌгаСНИіАыызАБЫсВаЛљЕФыФСД 46. ШЛКѓгывЛжжЩЬЦЗПЩЙКЕФСђЛЏЪдМСBTH ([Bu4N]2S6)дкЫЎШмвКжаЗДгІ, ПЩвдЗНБуЕижЦЕУДгЖўСђЧХЕНЮхСђЧХЕФЛЗыФ 47. Цфжа BTH ЕФгУСПМАЗДгІЪБМфВЛЭЌ, ПЩвдПижЦСђЧХЕФГЄЖШ(Eq. 4)[64].
6 КЌЯЉМќЛЗыФКЯГЩ
вбжЊЕФЗНЗЈжа, ЖрЪ§ЪЧНЋКЌЯЉМќЙЙМўв§ШыыФСД, ШЛКѓдйНјааЗжзгФкЙиЛЗИДЗжНт(RCM)ЗДгІЛђЗжзгФк HeckЗДгІ, ЪЕЯжЛЗКЯ
6.1 ЗжзгФк RCM ЗДгІЗНЪН
ИУЗНЪНЕФЯШОіЬѕМўЪЧЯШзщНЈыФСДЩЯСНИіЧХЭЗЧАЬхОљКЌЫЋМќЕФжаМфЬх. КѓепдкгаЛњюЩ(Ru)аЭЕФGrubbsЪдМСзїгУЯТЗЂЩњЗжзгФк RCM ЗДгІ, ЪЙыФСДЛЗЛЏ.
6.1.1 ЫЋВрСД O дзгЩЯЯЉБћУбЕФ RCM ЛЗКЯ
ыФСДЩЯЕФСНИі Ser, Thr Лђ Tyr ВаЛљЩЯЕФєЧЛљОЪЪЕБЕФзЊЛЏЗДгІ(Шч SN2, SNAr Лђ Mitsunobu ЗДгІ)в§ШыСНИіЯЉБћВрСД. ШЛКѓО Grubbs ЪдМСДпЛЏ, ЗЂЩњЗжзгФк RCM ЗДгІЩњГЩЯЉЛЗыФ(Scheme 45)[65].
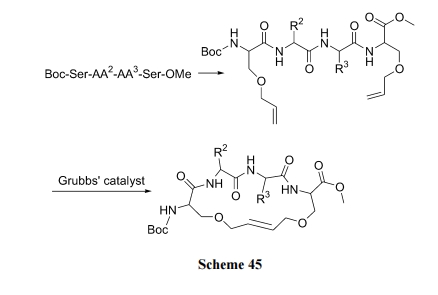
6.1.2 СНИіВаЛљ ІС-ЬМЩЯЯЉЮьЛљВрСДЕФ RCM ЛЗКЯ
ДгЯШдЄжЦЕФКЌЯЉБћЛљВрСДЕФАБЛљЫсЛђЦфбмЩњЮяЮЊЕЅЬх, ВЮгыыФСДзщзАКѓО Grubbs ЪдМСДпЛЏ, ЕУЕНЯргІЕФЯЉЛЗыФ[66](Eq. 5)
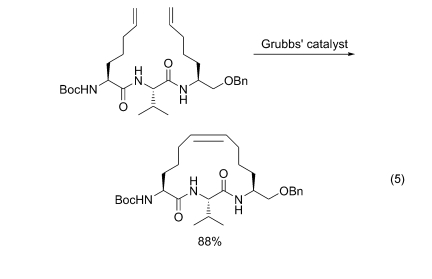
6.1.3 ыФСДжаСНИі N дзгЩЯЯЉЖЁЛљМфЕФ RCM ЛЗКЯ
ыФСДжаСНИі N дзгЩЯЯЉЖЁЛљМфЕФ RCM ЛЗКЯМћScheme 46[67].
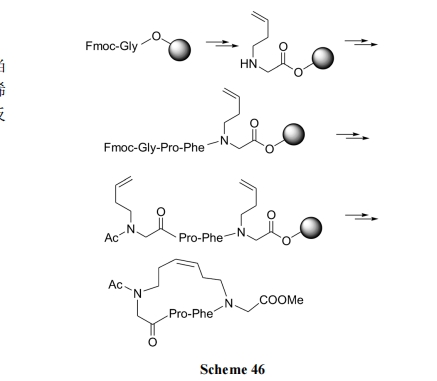
6.1.4 ФтыФСДЩЯВЛЖдГЦЫЋЯЉЕФ RCM ЛЗКЯ
вбНјШыСйДВЕФ HCV ЕААзУИвжжЦМС BILN-2061 ЪЧвЛжжФтыФЛЗКЯЕФЛЏКЯЮя. ЦфЛЗЩЯЕФЫЋМќЧХОЭЪЧдкВрСДИ§ЯЉгыВрСДЛЗМКЯЉжЎМфЕФ RCM ЗДгІаЮГЩЕФ, ДЫЗДгІЩњВњХњСПвбИпДя 400 kg вдЩЯ[68] (Scheme 47).

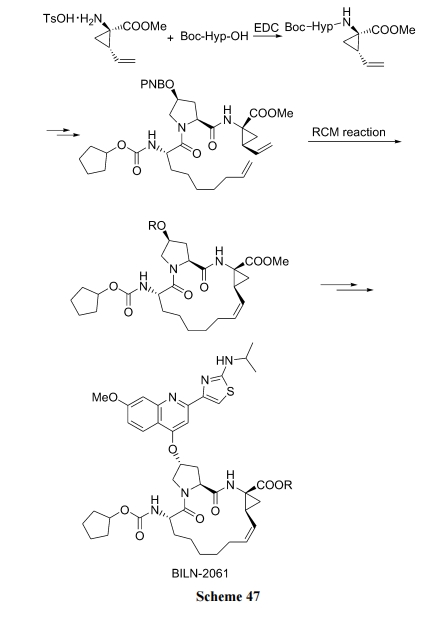
6.2 О Heck ЗДгІКЯГЩЯЉЛЗыФ
ЯЉЛђШВМќЩЯЕФВЛБЅКЭЬМдзгЗЂЩњЗМЬўШЁДњЪЧ HeckЗДгІЕФЬиЕу. вђДЫдкыФСДЕФВЛЭЌЮЛжУЗжБ№в§ШыЯЉ(ЛђШВ)МќМАЕтДњБН, зїЮЊКЯЛЗЕФЧАЬхНсЙЙ. ШЛКѓдкюйДпЛЏМСзїгУЯТ, ЗЂЩњЗжзгФк Heck ЗДгІ, ЩњГЩКЌЯЉ(ЛђШВ)ЧХЕФЛЗыФВњЮя.
ЯЉЖЁѕЃгыЖдМзѕЃЕтБНЕФМќКЯМћ Scheme 48[69]; ЯЉБћѕЃгыМфЕтДњмаАЗЕФМќКЯМћ Scheme 49[70].
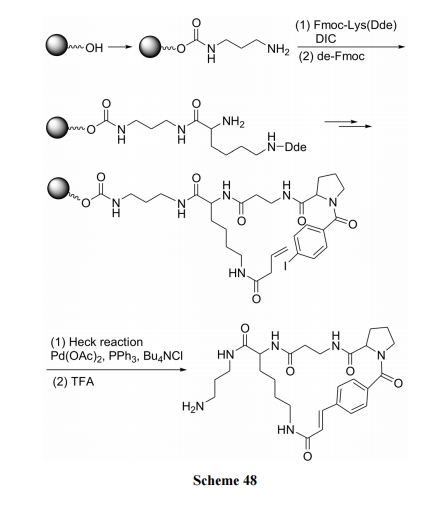
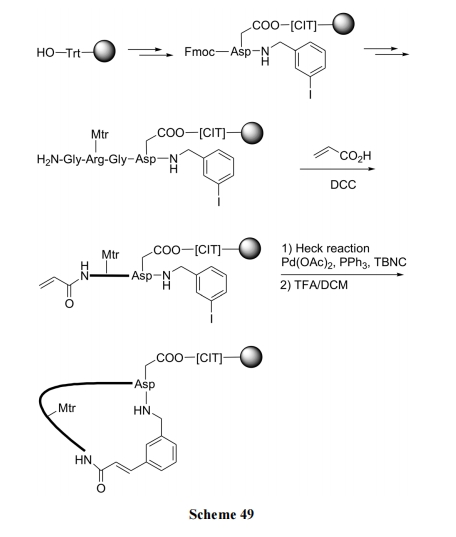
7 ИеадЧХЛЗыФКЯГЩ
7.1 Ш§ШЁДњЛЗЮьЭщЮЊЧХ
ЪзЯШНЋШ§ШЁДњЛЗЮьЭщЭЈЙ§єШЛљгыєЧЛљЪїжЌМќСЌ, ЫцКѓЯШКѓдкЛЅЮЊСкЮЛЕФєЧЛљМАЕўЕЊЩЯЗжБ№зщзАыФСДЕФ NЖЫЖЮМА C ЖЫЖЮ. зюКѓНјаа COOH гы NH2ЫѕКЯГЩЮЊКЌЛЗЮьЭщИеадЛЗЕФФкѕЃАЗЙиЛЗыФ[71] (Scheme 50).
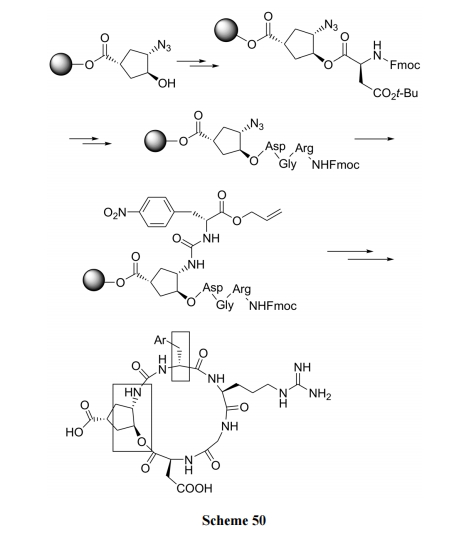
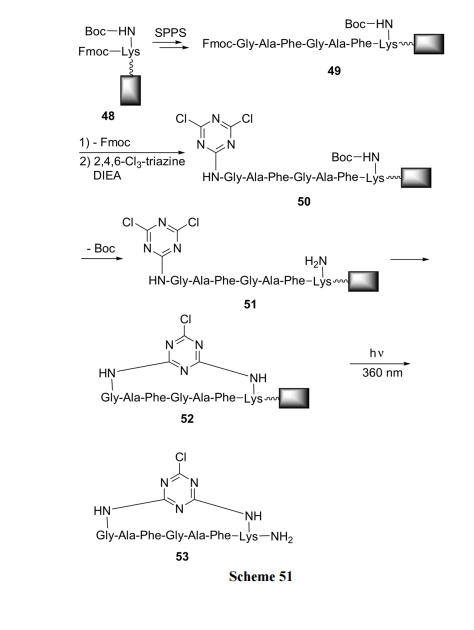
7.2 КЌШ§рКЛЗНсЙЙЕФЛЗыФ
гУе§НЛБЃЛЄЗНЪНЕФ Fmoc-Lys(Boc)зїЮЊзщзАыФСДМАЛЗЛЏЕФЛљДЁ. ЪзЯШНЋ Fmoc-Lys(Boc)ЭЈЙ§ЪЪЕБЕФМфИє(Spacer)НсЙЙгыЙЬЯрдиЬхСЌНгГЩЮЊ 48. ШЛКѓЭбГ§ Fmoc, НјаажБСДыФађСазщзАЕУЕНжаМфЬх 49. ЭбГ§ыФСД N ЖЫЕФFmoc жЎКѓ, НЋгЮРыЕФ NH2 гыШ§ТШШ§рКЩЯЕФвЛИіТШдзгНјаа SN2 ЗДгІ. ЫфШЛШ§ТШШ§рКЗжзгжаЕФ Cl ЗЧГЃЛюЦУ, ЕЋвђЙЬЯрдиЬхЕФМйЯЁЪЭаЇгІ, ЪЙШ§ТШШ§рКЗжзгжЛгавЛИіClгыыФЕФNH2ЗДгІ, ЕУЕН50. ЫцКѓЭбГ§РЕАБЫсВрСДЩЯЕФBoc, ЪЙNH2гЮРы, ГЩЮЊ51. дкМюЕФзїгУЯТ, Ш§рКЛЗЩЯЕФЕкЖўИі Cl дзггыРЕАБЫсВрСД NH2ЗЂЩњЗжзгФкЕФ SN2 ЗДгІ, ЩњГЩШ§рКЛЗЧЖШыЕФЛЗыФ 52[72] (Scheme 51).
7.3 ВЂЫФЧтпЛрЋЫЋЛЗНсЙЙЕФЛЗыФ
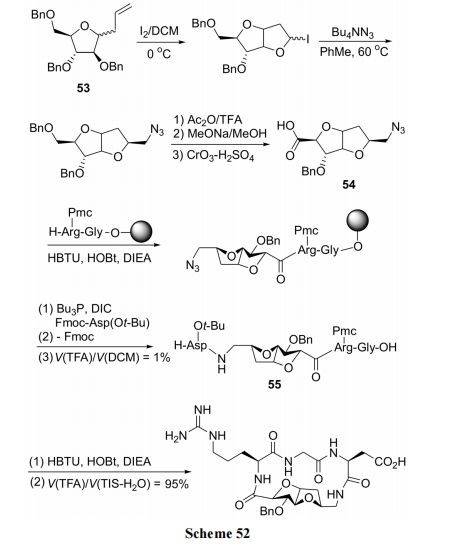
ЪзЯШАбИеадНсЙЙЕФЫЋВЂЫФЧтпЛрЋЯШгыыФСДНсКЯ, зюКѓгЩжЇМмСНВрЕФАБЛљЫсВаЛљЭъГЩФкѕЃАЗЛЗКЯ. ОпЬхКЯГЩвдЮьЬЧбмЩњЮя 53 ЮЊдСЯ, ОМИВНзЊЛЏЗДгІЕУЕНЖўВЂЫФЧтпЛрЋжЇМмаЭжаМфЬх 54. ЫцКѓгыЖўыФЪїжЌМќКЯ. дйНЋЖўВЂЫФЧтпЛрЋ N ЖЫЕФЕўЕЊЛљзЊЛЏЮЊ NH2, вдБугы FmocAsp(O-t-Bu)-OH ЫѕКЯ, жЦЕУЛЗЛЏЧАЬхНсЙЙ 55. зюКѓгУОЕфЕФыФЫѕКЯЬѕМўЭъГЩКЌ RGD ЦЌЖЮЕФЛЗыФжЦБИ(Scheme 52)[73].
7.4 ЖдЖўмаНщШыЕФЛЗыФ
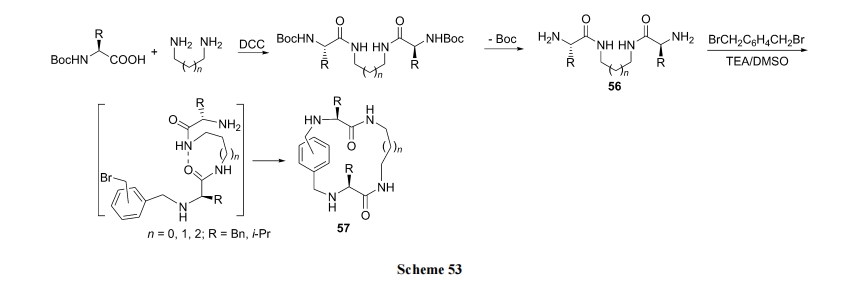
ЪзЯШвдЫЋБЖЮяжЪЕФСПЕФАБЛљЫсЖд ІС,Іи-ЖўАЗНјааЫЋЖЫѕЃЛЏ, МЬвдЭбГ§СНЖЫАБЛљЫсЩЯ N БЃЛЄЛљ, ЕУЕНЙиМќжаМфЬх56. ШЛКѓНјааЖдЮЛ(ЛђМфЮЛ)Жўфхмагы56ЕФСНИіNH2жЎМфЕФ SN2 ЗДгІ(ЗжСНВН), зюжеЕУЕНЖўмаНсЙЙЧЖШыЕФЫЋжйАЗЛЗыФ 57[74] (Scheme 53).
7.5 Н№ИеЭщНщШыЕФѕЅЛЗыФ
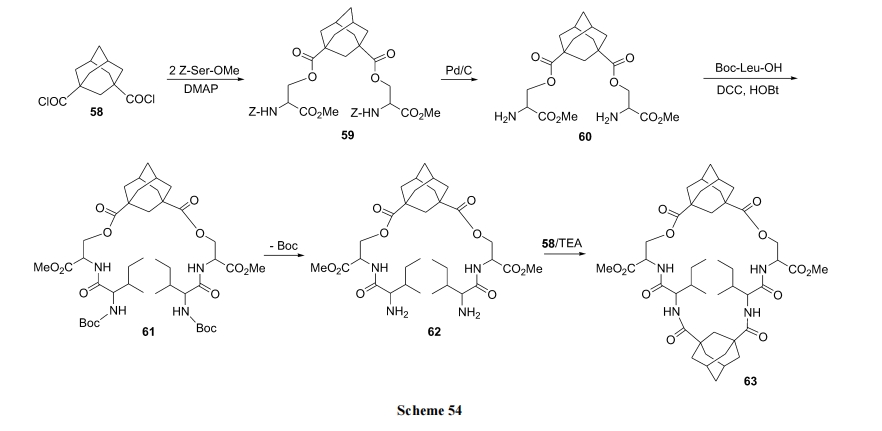
Н№ИеЭщНщШыЕФѕЅЛЗыФМћ Scheme 54[75].
8 жйЁЂЪхАЗЧХЛЗыФКЯГЩ
жйЁЂЪхАЗЧХЛЗыФКЯГЩжївЊгавдЯТШ§жжЗНЗЈ: ЪзЯШдЄжЦКЌжй(Ъх)АЗЕФжаМфЬх, КѓНјаажїСДЛЗКЯ[76,77]; ArF МАH2NAr жЎМфЕФМќКЯЙиЛЗ[78]вдМАфхввѕЃгыВрСДЩЯ NH2 ЕФМќКЯЙиЛЗ
9 Freidinger ОжВПЛЗыФ
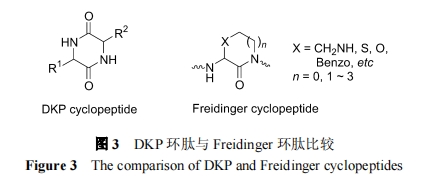
DKP ЪЧЭЗ-ЮВЯрСЌЕФФкѕЃАЗЛЗЖўыФЛЏКЯЮя. ЯрБШжЎЯТ, FreidingerЪЧвЛИіВаЛљЕФІС-ЬМдзггыгвВрЯрСкВаЛљЕФЕЊдзгЧХСЌЕФСэвЛРрЛЗЖўыФЛЏКЯЮя(ЭМ 3).СНепОљПЩдкыФСДжадіМгСЫИеадНсЙЙвђЫи, КѓепЕФНсЙЙЖрбљаддЖдЖГЌЙ§ DKP ЛЗыФ, вђДЫгаРћгкПЊЗЂНсЙЙЩЯИќЯёвЉ(drug like)ЕФЛюадЛЏКЯЮя.
КЯГЩ Freidinger ЛЗыФЕФЗНЗЈЗЧГЃЙуЗК, БОЮФзїепвбОЯЕЭГЕиНјааСЫЙщФЩ[80]. ДгКъЙлВпТдНВ, Freidinger ЛЗыФЕФКЯГЩЛљБОгЩ Scheme 55 ЫљЪО 4 жжТЗЯпЪЕЯж.

10 ЦфЫќНсЙЙЛЗыФКЯГЩ
10.1 СЊБНЧХЛЗыФ
гыЦеЭЈгаЛњаЁЗжзгСЊБНЛЏКЯЮяКЯГЩЯрЫЦ, ДЫРрЛЗыФЕФЛЗКЯЗНЗЈвВЪЧвдСНИіЕтДњБН(ЮЊЧХЭЗ)жЎМфЕФ Suzuki ЗДгІЁЂUllmann ЗДгІЛђбѕЛЏХМСЊЕШЗДгІЮЊЖрМћ[81].
10.2 Mannich МюЛЗыФ
ыФСДЩЯШчЙћКЌга Tyr ВаЛљ, ЦфЗгєЧЛљЕФ ІС-ЮЛПЩвдзїЮЊЛюЦУЧтЕФЙЉЬхгыАЗМАШЉЗЂЩњ Mannich ЫѕКЯ. ЪЕМЪЩЯыФСД N ЖЫАБЛљМДЪЧЯжГЩЕФАЗзщЗж, ЛЗКЯЪБжЛашЭтМгЕФШЉЙЙМўМДПЩжЦЕУ Mannich МюаЭЛЗыФ[82].
10.3 PNA ЛЗыФ
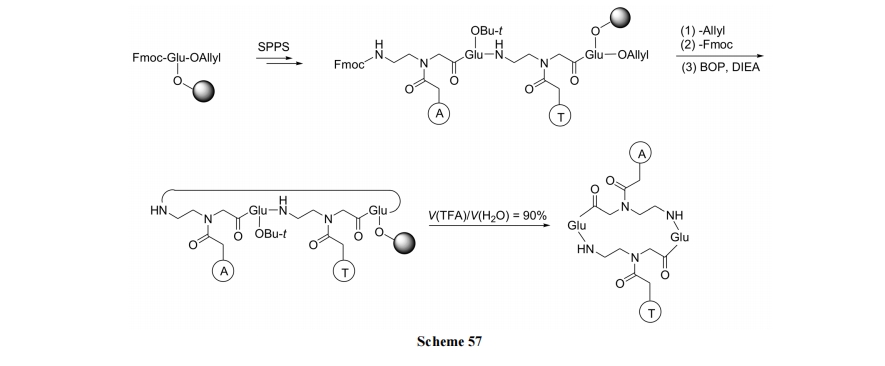
PNA ЪЧвдАБввЛљ-(N-МюЛљввѕЃ)-ИЪАБѕЃЮЊВаЛљДЎСЌаЮГЩЕФдгНЛыФ. ЦфЛЗЛЏВпТдПЩЗжЮЊЯШжЦБИЛЗыФСД, КѓМќСЌМю(Scheme 56)[83]МАЯШжЦБИКЌМюЛљЕФжБСДыФ, КѓЛЗКЯ(Scheme 57) [84]СНжж.
10.4 ЫЋ loop ЛЗыФ
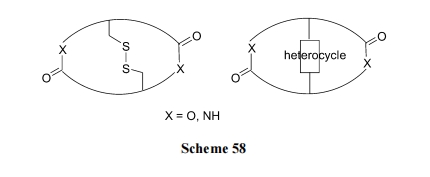
аэЖрЬьШЛВњЮяыФМАШЫЙЄКЯГЩЕФЯШЕМЛюадыФвдКЌгаСНИіСДЛЗ(loop)ЮЊНсЙЙЬиеї. етаЉЛЏКЯЮяЕФЧХСЌНсЙЙИїгаВЛЭЌ, дкЭЌвЛЗжзгФкСНИі loop жЎМфЙВгУЕФЧХНсЙЙ(ФкВрЧХ)гыЭтВрЧХЕФЧХНсЙЙЭљЭљЪЧВЛЯрЭЌЕФ. ЖрЪ§ЧщПіЯТ, ФкВрЧХгЩЖўСђМќЛђдгЛЗЙЙГЩ; ЭтВрЧХЭљЭљЪЧѕЃАЗМќЛђѕЅМќНсЙЙ(Scheme 58).
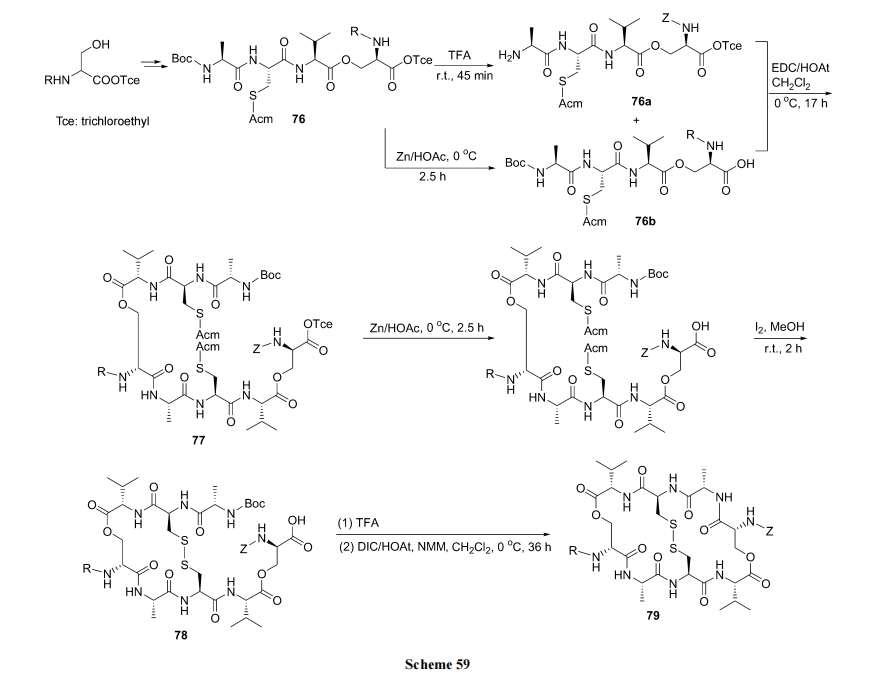
10.3.1 ЬьШЛВњЮяыФ Triostin A РрЫЦЮяЕФКЯГЩ
ЪзЯШжЦБИКЌ Cys ЕФШЋБЃЛЄѕЅЫФыФ 76. ШЛКѓЕФЗДгІЫГађЪЧСНИіЗжзгЕФ 76 НјааѕЃАЗМќЫѕКЯГЩЮЊ 77, ЗжзгФкаЮГЩЖўСђМќ(ФкВрЧХ)ЕУЕНжаМфЬх 78. зюКѓвЛЖдЧХЭЗаЮГЩѕЃАЗМќ(ЭтВрЧХ), ЭъГЩЫЋ loop 79 ЕФЙЙНЈ(Scheme 59)[85].
10.3.2 ЬьШЛВњЮяыФ[Ala7]-Phalloidin ЕФКЯГЩ
БОКЯГЩР§ФПБъЛЏКЯЮяЪЧвдпХпсЛЗЮЊФкВрЧХ, вдѕЃАЗМќЮЊЭтВрЧХЕФЫЋloopЛЗыФ. жївЊЕФКЯГЩВНжшЪЧЯШжЦБИКЌФкВрЧХЕФЦЌЖЮ, ШЛКѓбгГЄЭтВрСД. НгЯТРДЯШЪЙгвВрCONH ЙиЛЗ, дйаЮГЩзѓВрФкѕЃАЗМќ, ЙЙГЩЫЋ loop ВњЮя[86].
11 аЁНс
ЭГМЦБэУї, ОЁЙмжБСДыФЕФЪ§СПдЖдЖЖргкЛЗыФ, ЕЋФПЧАвбгаЕФыФРрЯШЕМЛЏКЯЮяЁЂгУгкСйДВЪдбщЕФыФРрКђбЁвЉМАвбОЩЯЪаЕФыФРрвЉЮяжа, ЛЗыФШДеМвЛАывдЩЯ. гЩДЫБэУї, дкаТвЉбаЗЂжа, ЛЗыФЕФГЩЙІТЪдЖдЖИпгкжБСДыФ. вђДЫ, ЩшМЦЁЂКЯГЩНсЙЙЖрбљЕФЛЗыФВЛЕЋНјвЛВНЗсИЛыФЛЏбЇЕФФкШн, ЖјЧвНЋЛсДйГЩИќЖрыФРраТвЉЕФГіЯж.
Утд№ЩљУїЃКБОЮФЮЊаавЕНЛСїбЇЯАЃЌАцШЈЙщдзїепМАддгжОЫљгаЃЌШчгаЧжШЈЃЌПЩСЊЯЕЩОГ§ЁЃЮФеТБъзЂгазїепМАЮФеТГіДІЃЌШчашдФЖСдЮФМАВЮПМЮФЯзЃЌПЩдФЖСддгжОЁЃ
